长QT 综合征发病机制研究新进展
2014-12-31李翠兰王震综述
李翠兰 王震 综述
(北京大学人民医院心脏中心,北京 100044)
1 概念及分子遗传学发现
长QT 综合征(long QT syndrome,LQTS)指具有心电图上QT 间期延长、T 波异常、易产生室性心律失常,尤其是尖端扭转型室性心动过速(TdP)、晕厥和猝死的一组综合征。迄今为止已发现的致病基因有20个,见表1[1-20]。
已知这种疾病的原因是患者从出生就携带了某些基因水平的变异,导致心肌细胞里一些细微的改变,虽然超声心动图显示心脏结构正常,但心脏的功能异常可在心电图上表现出来。目前已发现的20 个LQTS 致病基因中,KCNQ1 (LQT1)、KCNH2 (LQT2)及SCN5A(LQT3)为最常见的致病基因,约占遗传性LQTS 患者的80%。对患者进行基因检测时,发现已知的20 个基因突变的阳性检出率为80%~85%。也就是说,目前的技术水平还不能保证给所有的LQTS 患者检测出他们的致病基因,只有其中的80%~85%可以通过专门的检测机构获得他们确切的致病基因信息。
由于LQTS 的遗传方式多为常染色体显性遗传,所以在一个病人身上发现突变后,其突变遗传给后代的概率大约是50%。理论上讲,通过孕期的早期基因筛查还是可以检测出胎儿是否携带有其亲代的基因突变的,然后孕妇可以根据情况选择是否需要终止妊娠。只是限于各种原因,目前真正能够实施该项检测的机构还很少。
LQTS 中还有一种比较罕见的亚型同时伴有耳聋,称为JLN 综合征,是以两位最先发现该病的医生的名字命名的。这种有耳聋表型的LQTS 其患病率更低,约为百万分之一。致病基因为KCNQ1 和KCNE1(表1)。其遗传方式为常染色体隐性遗传,即父母双方各带一个相同或者不同的突变,然后同时把突变传给了子代。这种情况下子代的患病概率理论值为25%。由于患者携带两个突变的累加效应,通常这种亚型的患者临床症状更严重,发生致命性心脏事件的概率也更高。
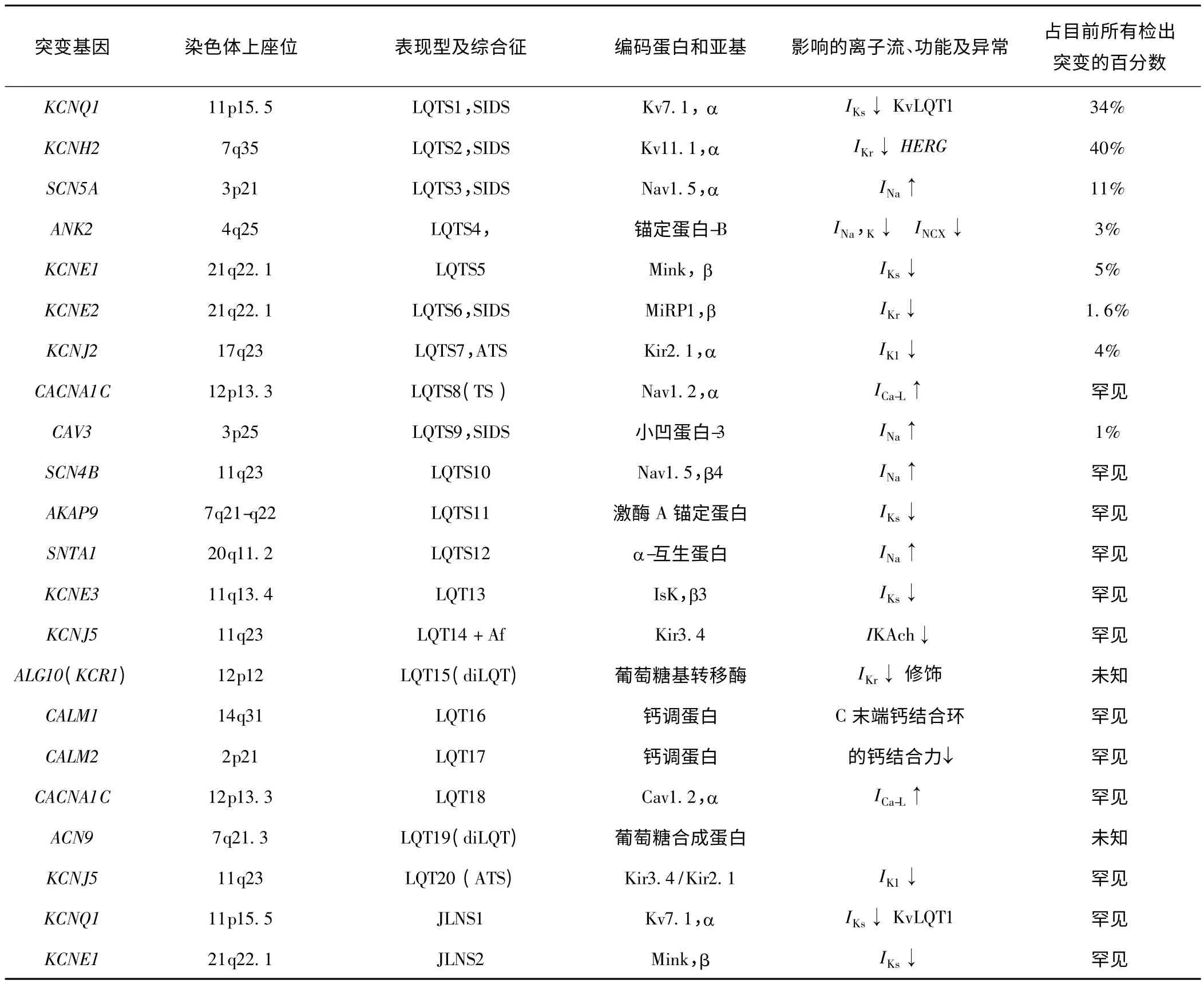
表1 长QT 综合征的分子遗传学
另外,药物引起的长QT 综合征(drug-induced LQT,diLQT)是临床上最常见的获得性LQTS。通常与抗心律失常药、抗组胺药和抗精神病药有关。这些药物被证明通过延长QT 间期,导致TdP,占所有处方量的2%~3%。大多数导致QT 间期延长的药物阻滞心肌细胞缓慢激活延迟整流钾电流(IKs),类似HERG 基因突变所导致的LQT2。1%~8%的患者接受QT 间期延长药物会表现出QT 间期延长或发展为TdP。因为QT 间期延长易感者容易出现快速室性心律失常如TdP 和心室颤动(Vf),所以该种心律失常患者的病死率可达10%~17%。因此diLQT 是过去几十年里已上市药物撤出市场的最常见原因。尽管这种不良反应在人群中相对少见(小于十万分之一),QT 间期延长也不总是诱发TdP。其他因素如心力衰竭、心室肥大、女性、低钾血症、隐性LQTS(存在基因突变而QT间期仍在正常范围)、猝死家族史等影响心脏的复极稳定性,也与药物诱发的TdP 有关。除引起遗传性LQTS 的致病基因外,最近还发现了2 个真正与diLQTS 有关的基因:ALG10B 和ACN9(见表1)。
在临床实践中,避免药物致QT 间期延长应注意以下几点:(1)不使用超过推荐剂量;(2)对已存在危险因素的患者减少使用剂量;(3)避免已知延长QT 间期的药物联合使用;(4)药物诱发TdP 的幸存患者和猝死者家族成员进行可能的基因筛查,是否存在隐性LQTS 等。
2 LQTS 基因检测原则
目前对LQTS 进行基因检测的专家共识推荐建议是[21]:(1)以下情况推荐进行LQT1~3(KCNQ1、KCNH2、SCN5A)的基因检测:基于病史、家族史及心电图表型(静息12 导联心电图和/或运动或儿茶酚胺应激试验)心脏病专家高度怀疑LQTS 的患者;无症状的特发性QT 间期延长者(其中青春前期QTc >0.48 s或成人QTc >0.50 s,排除继发性QT 间期延长因素,如电解质异常、药物因素、心肌肥厚、束支传导阻滞等)(Ⅰ类推荐)。(2)以下情况可考虑进行LQT1~3基因检测:无症状特发性QT 间期延长者,其中青春前期QTc >0.46 s,成人QTc >0.48 s(Ⅱb 类推荐)。(3)已在先证者发现LQTS 致病基因突变者,推荐其家族成员及相关亲属进行该特定突变的检测(Ⅰ类推荐)。(4)对药物诱发TdP 的先证者应考虑行基因检测(Ⅱb 类推荐)。(5)如果LQT1~3 突变检测阴性,但有QTc 间期延长,应考虑基因再评价,包括重复基因检测或进行其他更多致病基因检测(Ⅱb 类推荐)。
需要说明的是,以上专家推荐是从基因筛查所需的时间、费用等方面的投入产出性价比来评价的,随着基因测序技术的进步,现在新一代测序技术(靶标捕获与测序)从费用和实用性上都已走近了临床[22]。
3 LQTS 的主要亚型(LQT1~3)及其发病机制
3.1 LQT1 的致病原因
LQT1 的致病原因,在于KCNQ1 基因突变降低了心肌复极外向电流的主要成分IKs,使心室肌复极延迟,QT 间期延长。从经动脉灌注的犬心室肌楔形组织块标本LQT1 模型上观察到,IKs降低引起心外膜、中层(M 细胞)和心内膜细胞动作电位时程(APD)比较均一的延长,产生QT 间期延长和正常形态T 波,不伴显著的心室复极不均一性改变和心律失常。但当加入异丙肾上腺素后,跨壁不均一性增大,T 波形态改变,QT 间期进一步延长,出现TdP。这些结果可以解释LQT1 患者在安静状态时多显示正常形态T 波,大多无症状;而运动后交感兴奋可诱发双峰T 波,运动后早期出现一过性宽大T 波,心室复极不均一性增大,QTc 延长。LQT1 患者心脏事件多发生在剧烈运动中或运动后。从理论上讲,β-肾上腺素能刺激可以增加IKs、L型钙电流(ICa-L)和氯离子流(ICl(Ca)及ICl-cAMP),其总效应为复极外向电流增加和APD 缩短,因而正常人在剧烈运动时心率加快,QTc 缩短。但当KCNQ1 基因突变造成IKs离子通道蛋白结构异常时,IKs通道不再受β-肾上腺素能刺激所支配,心室复极离子流的总效应变为内向电流增加,外向电流减少,APD 延长,引起后除极,甚至诱发TdP 和Vf,以至猝死。治疗量的β受体阻滞剂可防止运动中的双峰T 波和运动后的宽大T波,避免心室复极不均一性增大和运动引起的QTc 进一步延长,从而起到了预防致命性快速室性心律失常的作用。多年来的实践证明,治疗量的β受体阻滞剂对绝大多数的LQT1 患者有保护作用。
3.2 LQT2 由心脏钾通道基因HERG(又称KCNH2)突变引起
KCNH2 基因编码一个具有6 个跨膜结构域的心脏K+通道的α 亚基。KCNH2 与MinK 的同源蛋白MiRP1(由KCNE2 基因编码)组成一个有功能的离子通道,在心脏中产生快速激活延迟整流钾电流(IKr)。电流主要在平台期或随后的3 相快速复极相期起作用。LQT2 是由于HERG 突变降低了心肌复极外向电流IKr,使心室肌复极延迟,QTc 延长。大多数HERG突变引起HERG 蛋白在细胞内的转运障碍,使之无法在膜上表达或表达缺陷,导致IKr离子流减低。
在犬心室肌楔形组织块标本LQT2 模型上观察到,降低的IKr引起心外膜、中层(M 细胞)和心内膜细胞APD 不均一性延长,以中层(M 细胞)为主,跨壁不均一性增大。尤其在低钾的条件下,心电图上产生双峰T 波,再介入异丙肾上腺素,可诱发TdP。与LQT1有别,LQT2 患者在安静状态下呈现的双峰或切迹型T波反映了其心肌复极的不均一性,惊吓或情绪激动往往诱发心脏事件。有人发现补钾疗法可使T 波切迹变浅、心室复极不均一性降低、QTc 缩短。但其保护作用还有待于大样本资料积累后评估。IKr离子通道对药物敏感,是绝大多数获得性LQTS 的靶通道。
3.3 LQT3 由SCN5A 突变引起
典型的LQT3 是由hNav1.5(SCN5A)突变造成晚钠电流(INa-L)反复开放,延缓电流衰退,使AP 平台期延长。SCN5A 突变通过功能放大(gain-of-function)机制(突变通道的功能正常,但特性改变了)引起LQT3。这种变化在慢频率下尤其明显,可以解释LQT3 患者存在心动过缓依赖性ST 段延长、晚发T 波和QTc 延长。LQT3 的心脏事件多发生于睡眠或安静状态下。与LQT1 相反,心率加快可减少INa-L,使APD 缩短,T波和QTc 正常化。
4 从LQTS 致病机制入手探索基因特异性治疗
从LQTS 第一个致病基因发现起,研究者就一直希望通过对各个基因致病机制的透彻理解来找到纠正治疗的方法。但这些遗传疾病致病机制的多样性和复杂性使得目前所有的方法离应用于病人还差得很远。在3 个主要亚型中,目前研究最多的是LQT2。下面就以引起LQT2 的致病基因KCNH2 为例汇总一下近年来的新成果以及潜在的治疗启示。
相对于我们熟知的负显性机制、单倍体不足机制,大多数HERG 突变是引起HERG 蛋白在细胞内的转运障碍,使之无法在膜上表达或表达缺陷,导致IKr离子流减低[23],又称之为2 类机制。但不同位点或区域的突变会有一些特殊的作用方式。比如,Gong等[24]的研究表明,LQT2 的无义突变如W1001X 和R1014X,会通过一种称作无义介导的mRNA 衰减(nonsense-mediated mRNA decay,NMD)机制引起成熟突变RNA 水平的下降,而不是产生截短蛋白。提示通过NMD 导致成熟hERG 突变,mRNA 降解是LQT2 病人无义或移码突变的重要致病机制。随后的进一步研究发现,用UPF1 进行RNA 介导的基因敲减抑制NMD 后会增加Q1070X 突变通道蛋白的表达及hERG的电流幅度。更重要的是,发现用反义吗啉寡聚核苷酸特异抑制下游内含子剪接位点后可以消除Q1070X突变mRNA 的NMD 作用,恢复功能性Q1070X 突变通道的表达。在LQT2 移码突变中也观察到了这种反义吗啉寡聚核苷酸恢复突变通道功能表达的现象。提示用反义吗啉寡聚核苷酸抑制NMD 可能是某些携带无义和移码突变的LQT2 病人潜在的治疗手段[25]。2014 年初刚刚发表的研究又发现,NMD 是否发挥作用与无义突变引进的提前终止密码子(premature termination codon,PTC)的位置有关,即PTC 必须位于3'端最后两个外显子连接处上游至少54~60 个核苷酸,而且下游的内含子还必须功能正常,因为用反义吗啉寡聚核苷酸抑制下游内含子剪切后可抑制NMD,挽救突变的功能表达[26]。
位于N 末端的突变又是另外一种情况。Q81X 无义突变对NMD 机制不敏感,其翻译在Met124位置重新开始[27],导致产生N 端截短的hERG 蛋白通道,门控特性发生改变。C39X、C44X 突变的翻译则在Met60重新开始,这样其前面的59 个氨基酸残基的缺失几乎占到PAS 域(信号蛋白的一种结构域名称)的1/3,引起的后果是蛋白转运障碍,hERG 电流完全丧失。PAS域的部分缺失还可引起突变通道蛋白的加速降解。突变和野生型共表达实验证实这几个突变不影响野生型通道的功能。提示通过翻译重新启动机制产生截短蛋白转运障碍和通道功能异常,是部分携带靠近编码序列N 端PTC 突变的LQT2 患者的致病机制。还有,Ying 等研究了PAS 域的突变对该区域的空间折叠以及PAS 域和通道其他部分相互关系的影响。结果发现,除R56Q 外的所有其他PAS 区域突变都会破坏PAS 域的热稳定性。位于PAS 域表面疏水区块的6 个突变还会影响PAS 域和N 端截短hERG 蛋白通道的结合[28]。相反,另外4 个表面突变(C64Y、T65P、A78P、I96T)和1 个埋在蛋白内部的突变(L86R)并不影响PAS 域与截短通道的结合。这个研究强调了PAS 域和hERG 通道其余部分之相互作用在通道组装过程中的重要作用,这也解释了携带PAS 区域突变的患者其转运障碍比之携带非折叠区突变的要严重。
还有一类影响到9 号内含子的突变又是另外一种机制。KCNH2 编码Kv11.1 钾通道,负责心脏IKr。其中编码蛋白有两种异构体形式:全长的Kv11.1a 和C末端截短的Kv11.1a-USO,它们的表达比例在通道功能的调节中起重要作用。那么C 末端截短形式的异构体是怎么形成的呢?这是由9 号内含子上剪切位点和备择polyA 之间相互竞争来决定的。Gong 等[29]首先在一个很大的LQTS 家系发现了一个新的KCNH2突变IVS9-2delA,这是一个在9 号内含子上3'受体位置的核苷酸AG 缺失了一个A。然后研究者设计了一个包含9 号内含子的全长KCNH2 基因结构,来研究突变对Kv11.1a 和Kv11.1a-USO 两种异构体在mRNA、蛋白和功能水平表达比例的影响。结果发现,这个突变会破坏9 号内含子的正常剪切并造成一种独特的polyA 结构,在HEK293 细胞和HL-1 心肌细胞可以引起有功能的Kv11.1a 向无功能的Kv11.1a-USO 异构体的转换。同时在病人淋巴细胞分离得到的mRNA上也证明IVS9-2delA 引起了异构体的转换。这项研究提示,IVS9-2delA 突变引起了功能性的Kv11.1a 异构体向非功能性的Kv11.1a-USO 异构体的表达转换。这种Kv11.1 异构体转换代表了一种新的LQTS 致病机制。该研究组的另一项研究[30]随后发现,KCNH2 9号内含子polyA 的信号活性有赖于顺式作用元件(cisacting elements)正常。这些元件发生突变后就会降低Kv11.1a-USO 表达,增加Kv11.1a mRNA 表达和蛋白翻译,增大通道电流。更重要的是,用反义吗啉寡聚核苷酸阻滞这些元件后,KCNH2 9 号内含子的polyA备择过程就被转换成正常剪接途径,导致主要产生Kv11.1a,其电流显著增加。提示Kv11.1a 异构体的表达可被反义抑制方式上调,KCNH2 内含子polyA 的反义抑制代表了一种新的增加Kv11.1 通道功能的方法。
在基于发病机制的基因治疗方法探讨方面,除上面提到过的用UPF1 进行RNA 介导的基因敲减或用反义吗啉寡聚核苷酸抑制NMD 的方法外,有研究[31]利用等位基因特异的RNA 干扰(RNAi)技术将突变基因敲减后,使从LQT2 病人诱导多能干细胞分化来的心肌细胞功能重新恢复了正常,提示这个方法对常染色体显性遗传疾病中的负显性突变是一个潜在的新的治疗途径。
[1]Wang Q,Curran ME,Splawski I,et al.Positional cloning of a novel potassium channel gene:KVLQT1 mutations cause cardiac arrhythmias[J].Nat Genet,1996,12(1):17.
[2]Wang Q,Shen J,Splawski I,et al.SCN5A mutations associated with an inherited cardiac arrhythmia,long QT syndrome[J].Cell,1995,80(5):805.
[3]Mohler PJ,Schott JJ,Gramolini AO,et al.Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death[J].Nature,2003,421(6923):634-639.
[4]Splawski I,Tristani-Firouzi M,Lehmann MH,et al.Mutations in the hminK gene cause long QT syndrome and suppress IKs function[J].Nat Genet,1997,17(3):338.
[5]Abbott GW,Sesti F,Splawski I,et al.MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia[J].Cell,1999,97(2):175.
[6]Splawski I,Shen JX,Timothy KW,et al.Spectrum of mutation in long QT syndrome gene KvLQT1,HERG,SCN5A,KCNE1,and KCNE2[J].Circulation,2000,102:1178.
[7]Plaster NM,Tawil R,Tristani-Firouzi M,et al.Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome[J].Cell,2001,105(4):511-519.
[8]Splawski I,Timothy KW,Sharpe LM,et al.Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism[J].Cell,2004,119(1):19-31.
[9]Vatta M,Ackerman MJ,Ye B,et al.Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome[J].Circulation,2006,114(20):2104-2112.
[10]Medeiros-Domingo A,Kaku T,Tester DJ,et al.SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome[J].Circulation,2007,116(2):134-142.
[11]Chen L,Marquardt ML,Tester DJ,et al.Mutation of an A-kinase-anchoring protein causes long-QT syndrome[J].Proc Natl Acad Sci U S A,2007,104(52):20990-20995.
[12]Ueda K,Valdivia C,Medeiros-Domingo A,et al.Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex[J].Proc Natl Acad Sci U S A,2008,105(27):9355-9360.
[13]Ohno S,Toyoda F,Zankov DP,et al.Novel KCNE3 mutation reduces repolarizing potassium current and associated with long QT syndrome[J].Hum Mutat,2009,30(4):557-563.
[14]Yang Y,Yang Y,Liang B,et al.Identification of a Kir3.4 mutation in congenital long QT syndrome[J].Am J Hum Genet,2010,86(6):872-880.
[15]Hayashi K,Fujino N,Ino H,et al.A KCR1 variant implicated in susceptibili-ty to the long QT syndrome[J].J Mol Cell Cardiol,2011,50(1):50-57.
[16]Petersen CI,McFarland TR,Stepanovic SZ,et al.In vivo identification of genes that modify ether-a-go-go-related gene activity in Caenorhabditis elegans may also affect human cardiac arrhythmia[J].Proc Natl Acad Sci U S A,2004,101(32):11773-11778.
[17]Crotti L,Johnson CN,Graf E,et al.Calmodulin mutations associated with recurrent cardiac arrest in infants[J].Circulation,2013,127(9):1009-1017.
[18]Boczek NJ,Best JM,Tester DJ,et al.Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel,CACNA1C,linked to autosomal dominant long QT syndrome[J].Circ Cardiovasc Genet,2013,6(3):279-289.
[19]Weeke P,Mosley JD,Hanna D,et al.Exome sequencing implicates an increased burden of rare potassium channel variants in the risk of drug-induced long QT interval syndrome[J].J Am Coll Cardiol,2014,63(14):1430-1437.
[20]Kokunai Y,Nakata T,Furuta M,et al.A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2.1[J].Neurology,2014,82(12):1058-1064.
[21]中华医学会心血管病学分会,中华心血管病杂志编辑委员会.遗传性心脏离子通道病与心肌病基因检测中国专家共识[J].中华心血管病杂志,2011,39(12):1073-1082.
[22]Rehm HL.Disease-targeted sequencing:a cornerstone in the clinic[J].Nat Rev Genet,2013,14(4):295-300.
[23]Anderson CL,Delisle BP,Anson BD,et al.Most LQT2 mutations reduce Kv11.1 (hERG)current by a class 2 (trafficking-deficient)mechanism[J].Circulation,2006,113(3):365-373.
[24]Gong Q,Zhang L,Vincent GM,et al.Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome[J].Circulation,2007,116(1):17-24.
[25]Gong Q,Stump MR,Zhou Z.Inhibition of nonsense-mediated mRNA decay by antisense morpholino oligonucleotides restores functional expression of hERG nonsense and frameshift mutations in long-QT syndrome[J].J Mol Cell Cardiol,2011,50(1):223-229.
[26]Gong Q,Stump MR,Zhou Z.Position of premature termination codons determines susceptibility of hERG mutations to nonsense-mediated mRNA decay in long QT syndrome[J].Gene,2014,539(2):190-197.
[27]Stump MR,Gong Q,Zhou Z.LQT2 nonsense mutations generate trafficking defective NH2-terminally truncated channels by the reinitiation of translation[J].Am J Physiol Heart Circ Physiol,2013,305(9):H1397-H1404.
[28]Stump MR,Gong Q,Packer JD,et al.Early LQT2 nonsense mutation generates N-terminally truncated hERG channels with altered gating properties by the reinitiation of translation[J].J Mol Cell Cardiol,2012,53(5):725-733.
[29]Gong Q,Stump MR,Deng V,et al.Identification of Kv11.1 isoform switch as a novel pathogenic mechanism of long QT syndrome[J].Circ Cardiovasc Genet,2014,Jul 15[Epub ahead of print].
[30]Gong Q,Stump MR,Zhou Z.Upregulation of functional Kv11.1 isoform expression by inhibition of intronic polyadenylation with antisense morpholino oligonucleotides[J].J Mol Cell Cardiol,2014,Aug 14.pii:S0022-2828(14)00254-5[Epub ahead of print].
[31]Matsa E,Dixon JE,Medway C,et al.Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes[J].Eur Heart J,2014,35(16):1078-1087.
