液相色谱-串联质谱法检测牛奶中β-内酰胺类药物残留的研究
2014-12-21文∕贾涛
文∕贾 涛
(北京市饲料监察所)
1 方法原理
牛奶中β-内酰胺类药物经乙腈提取,正己烷脱脂后,经C18净化小柱净化,取适量上清液过0.22 µm滤膜后,供液相色谱-串联质谱仪测定。
2 试剂和材料
以下所用的试剂,除特别注明者外均为分析纯试剂;水为符合GB/T 6682规定的二级水。
青霉素G、青霉素V、阿莫西林、羧苄西林、氨苄西林、苯唑西林、氯唑西林、萘夫西林、头孢喹肟、头孢氨苄、头孢拉定、头孢唑啉和头孢哌酮对照品(纯度均大于95.0%);乙腈(色谱纯);正己烷;甲酸(色谱纯)。
标准储备液(1 mg/mL):准确称取适量的青霉素G、青霉素V、阿莫西林、羧苄西林、氨苄西林、苯唑西林、氯唑西林、萘夫西林、头孢喹肟、头孢氨苄、头孢拉定、头孢唑啉和头孢哌酮,用50%乙腈水溶液溶解稀释,分别配制成1 mg/mL的标准储备液。4 ℃下保存,有效期为1 周。
混合标准工作液(10 µg/mL):分别准确吸取0.1 mL的青霉素G、青霉素V、阿莫西林、羧苄西林、氨苄西林、苯唑西林、氯唑西林、萘夫西林、头孢喹肟、头孢氨苄、头孢拉定、头孢唑啉和头孢哌酮标准储备液至10 mL容量瓶中,用水稀释至刻度,混匀即得。4 ℃下保存,有效期为1 周。
3 仪器和设备
超高效液相色谱-串联质谱仪(配电喷雾离子源,美国Waters公司,Waters Quattro Premier XE);分析天平(感量0.00001 g);天平(感量0.01 g);高速离心机;涡旋混合器;水平振荡器;固相萃取装置;BakerBond C18固相萃取柱(500 mg/6 mL,或相当者);氮吹仪;滤膜(0.22 µm)。
4 试验方法
4.1 试验仪器条件
4.1.1 液相色谱条件
色谱柱:BEH C18(50 mm×2.1 mm,1.7 µm);流动相:A相为0.1%甲酸乙腈溶液,B相为0.1%甲酸水溶液;梯度洗脱:0~1 min,维持5%A;1~2.5 min,5%A线性变化至50%;2.5~4 min,维持50%A;4~5 min,维持5%A。流速:0.3 mL/min;柱温:30 ℃;进样量:10 µL。
4.1.2 质谱条件
离子源:电喷雾离子源;扫描方式:正离子扫描;检测方式:多反应监测;电离电压:3.2 kV;源温:110 ℃;雾化温度:350 ℃;锥孔气流速:50 L/h;雾化气流速:650 L/h;测试药物定性、定量离子对及对应的锥孔电压、碰撞能量见表1。
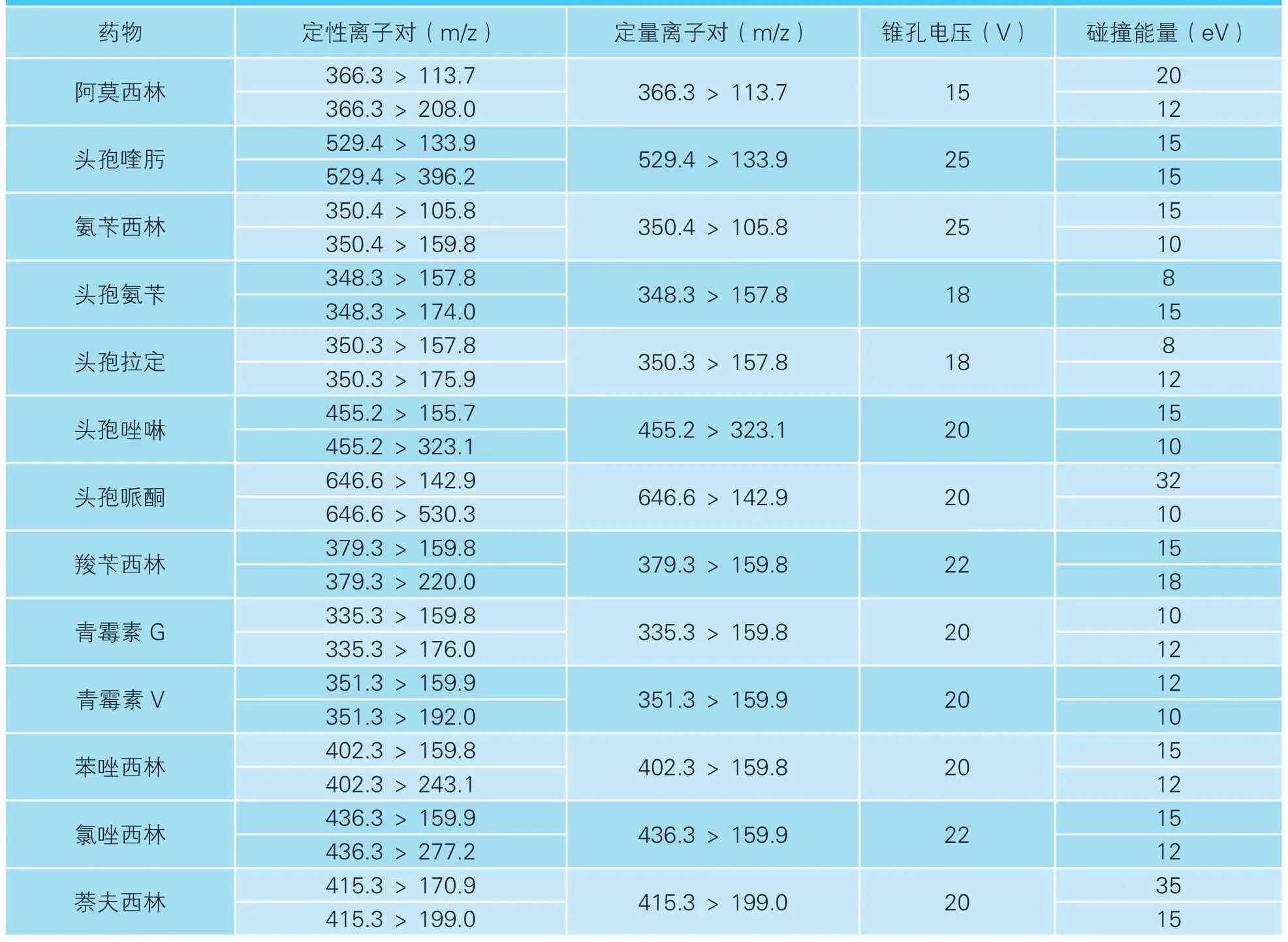
表1 13 种β-内酰胺类药物定性、定量离子对及锥孔电压、碰撞能量
4.2 样品前处理
4.2.1 提取
称取(2±0.02)g牛奶样品,置于50 mL离心管内,加乙腈8 mL,涡旋混合后,中速振荡5 min,10 000 r/min离心10 min。取上清液于另一个50 mL离心管内,加正己烷5 mL,涡旋混合后中速振荡5 min,5 000 r/min离心5 min,弃上层有机相,下层溶液作为备用液。
4.2.2 净化
C18小柱依次用乙腈5 mL、水10 mL活化,取全部备用液过柱同时收集于15 mL玻璃试管内,挤干,于40 ℃下氮气吹至体积小于1 mL。加水定容至1 mL,充分涡旋混匀,转移至1.5 mL塑料离心管内,15 000 r/min离心10 min,取适量上清液过0.22 µm滤膜后,供液相色谱-串联质谱仪测定。
4.2.3 测定法
取试料溶液和基质匹配标准溶液,作单点或多点校准,外标法计算。试料溶液和基质匹配标准溶液中青霉素G、青霉素V、阿莫西林、羧苄西林、氨苄西林、苯唑西林、氯唑西林、萘夫西林、头孢喹肟、头孢氨苄、头孢拉定、头孢唑啉和头孢哌酮的特征离子质量色谱峰面积均应在仪器检测的线性范围之内。试料溶液中的离子相对丰度与基质匹配标准溶液中的离子相对丰度相比,符合表2的要求。
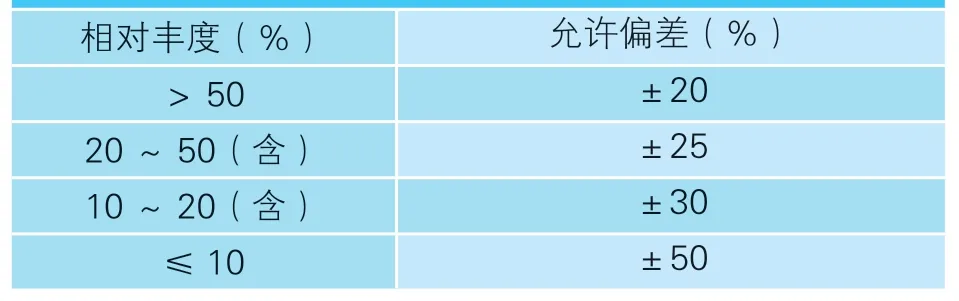
表2 试料溶液中离子相对丰度的允许偏差范围
4.2.4 结果计算和表述
或基质匹配标准曲线校准:由As=aCs+b,
式中:
X——供试试料中β-内酰胺类药物残留量(µg/kg);
Cs——基质匹配溶液中相应β-内酰胺类药物浓度(ng/mL);
C——供试试料溶液中相应β-内酰胺类药物浓度(ng/mL);
As——基质匹配溶液中相应β-内酰胺类药物峰面积;
A——供试试料溶液中相应β-内酰胺类药物峰面积;
V——浓缩后定容体积(mL);
m——供试试料质量(g)。
注:计算结果需扣除空白值,测定结果用平行测定的算术平均值表示,保留三位有效数字。
4.3 线性范围
分别准确量取13 种β-内酰胺类药物系列混合标准溶液0.1 mL,依次加入空白组织经提取、净化及浓缩后的溶液中,加水定容至1 mL,充分混匀,制得浓度为5、10、50、100、200和500 ng/mL的基质匹配系列混合标准溶液,离心过滤后上机测定。以特征离子质量色谱峰面积为纵坐标,标准溶液浓度为横坐标,绘制标准曲线。空白牛奶基质匹配得到的回归方程及相关系数(R2)见表3。从中可以看出:13 种β-内酰胺类药物在5~500 ng/mL的浓度范围内呈现良好的线性关系,R2均大于0.990。

表3 13 种β-内酰胺类药物回归方程及相关系数(R2)
4.4 方法的灵敏度
空白试样按相同的步骤处理后进行测定,测定结果表明:在相应的保留时间,空白试样对所测分析物无干扰。检测限(LOD)与定量限(LOQ):添加适量混合标准溶液于2 g空白样品,经提取净化后测定,根据所测药物的S/N>3(按PtP算)为方法检测限,S/N>10(按PtP算)为方法定量限的原则,测得青霉素G、青霉素V、阿莫西林、羧苄西林、氨苄西林、苯唑西林、氯唑西林、萘夫西林、头孢喹肟、头孢氨苄、头孢拉定、头孢唑啉和头孢哌酮13 种药物在牛奶中的检测限为1 µg/kg,定量限为2 µg/kg。
4.6 检测方法的准确度和精密度考察
采用标准添加法,在空白牛奶中添加低、中、高3个不同浓度的13 种β-内酰胺类药物进行回收率试验,各浓度进行5 个样品平行试验,重复3次,求批内、批间相对标准偏差,汇总结果见表4。从表中试验结果可以看出,本方法在空白牛奶中进行2~300 ng/g添加浓度检测,方法的回收率为70%~120%,批内、批间相对标准偏差均小于20%。

表4 空白牛奶中13 种β-内酰胺类药物添加回收率试验(n=5)
5 结果与讨论
5.1 线性范围
空白基质中13 种β-内酰胺类药物在5~500 ng/mL浓度范围内线性相关,相关系数均大于0.990。
5.2 灵敏度
青霉素G、青霉素V、阿莫西林、羧苄西林、氨苄西林、苯唑西林、氯唑西林、萘夫西林、头孢喹肟、头孢氨苄、头孢拉定、头孢唑啉和头孢哌酮在牛奶中的检测限为1 µg/kg,定量限为2 µg/kg。
5.3 精确度
在牛奶添加浓度为2~50 µg/kg范围内,本方法的回收率为70%~120%;批内、批间RSD均小于20%。
5.4 β-内酰胺类药物的残留情况与研究检测方法的意义
β-内酰胺类药物(β-lactams)具有自然界中鲜见的β-内酰胺基母核,其中发展迅速、品种较多的两类是青霉素类(penicillins,PENs)和头孢菌素类(cephalosporins,又称先锋霉素)。常见的青霉素类药物包括天然的青霉素G(又称苄青霉素)等以及半合成的阿莫西林、氨苄西林和苯唑西林等;头孢菌素类药物较青霉素类稳定,具有过敏反应低,杀菌力强等优势,该类药物品种繁较多,不断更新,典型的有头孢氨苄、头孢喹肟、头孢唑啉等。
β-内酰胺类药物在医学和兽医上都是重要的一类抗菌药物,应用十分广泛。由于过敏反应及细菌产生耐药性等原因,许多国家都对动物使用该类药物以及在动物源食品中的残留情况进行了严格监控。欧盟、美国和我国等多个国家都制定了牛奶等食品中的最高残留限量。β-内酰胺类药物残留分析方法已有大量报道,主要有液相色谱法(HPLC)法、免疫分析法和微生物测定法等,本方法采用快速简便、专属性强、灵敏度高的液相色谱-串联质谱法(LC-MS/MS)法作为该类药物的确证方法,以保障对该类兽药的残留监控。
5.5 前处理条件的确定
对于β-内酰胺类药物的提取大多使用水和有机溶剂进行提取,有时还要加入钨酸钠、硫酸等无机酸来沉淀蛋白质,但是一些天然的药物,如青霉素G等不耐酸,在酸性条件下又易分解,因此最终试验中采用了1∶4的水和乙腈进行提取的方法。为降低背景杂质对分析物的干扰,常需做进一步的净化,常用的固相萃取柱为C18柱和HLB柱,本试验比较了Sep-pak、Supelco、Varian、BakerBond C18小柱和HLB小柱,结果发现使用填料致密的BakerBond C18小柱去除杂质、净化样品的效果最好。
5.6 离子对的确定
根据欧盟2002/657/EC要求,要确证β-内酰胺类药物至少需要3 个识别点(IP),试验中分别选择了母离子以及对应的2 个响应较强的子离子作为定性依据,这样,1 个母离子有1个IP点,对应的2 个子离子分别有1.5 个IP点,达到了4 个IP点,很好地满足了欧盟有关法规的要求。
6 结论
通过线性范围试验、回收率试验等认为,液相色谱-串联质谱法检测速度较快、定性定量准确、回收率较高,完全能满足13 种β-内酰胺类药物在牛奶中残留检测方法的要求。
