壳聚糖聚乳酸羟基乙酸纳米粒子作为抗瘤药物载体的研究*
2014-12-02马方奎包子娴陈西广
马方奎,包子娴,陈西广
(中国海洋大学海洋生命学院,山东 青岛 266003)
近年来,阿霉素作为广谱抗肿瘤药物,常用于多种癌症的治疗,其对乳腺癌也具有很好的治疗效果[1],但是由于肿瘤多药耐药性的存在,影响了阿霉素的抗肿瘤效果。目前针对耐药肿瘤细胞系来研究解决多药耐药性的报道已经很多,而纳米靶向载体成为克服肿瘤多药耐药性和提高药物靶向的一个重要途径。Bae等[2]的研究表明,通过纳米载体包载阿霉素进行药物释放,可以显著降低阿霉素的毒性和副作用。
聚乳酸羟基乙酸(PLGA)作为FDA认可的生物医用材料已经被用于大量的临床试验,已经成功用于多种抗肿瘤化疗药物(尤其是疏水药物)的载体进行给药。De等[3]研究利用PLGA包载抗癌药物紫杉醇,可以显著增加药物在4T1肿瘤细胞中的摄取和抑制作用。然而PLGA包载的纳米载药粒子被细胞吞噬系统的快速吞噬,很大程度上影响了PLGA载药纳米粒子的靶向效果。因此PLGA纳米载药运输的最大的问题是如何有效规避网状内皮系统的吞噬(Reticuloendothelial system,RES)。而这可以通过亲水聚合物的表面修饰来改变纳米粒子表面的亲水性,从而降低纳米粒子表面对调理素的吸收,而且减少巨噬细胞的吞噬,提高载药的靶向性[4]。利用壳聚糖修饰PLGA纳米粒子后,进一步用亲水性的PEG包被修饰来改变纳米粒子表面的亲水性,以降低细胞表面对调理素的吸收从而提高靶向性。Hu等[5]报道,无毒且生物相容性好的阳离子聚合物壳聚糖可以促进纳米粒子穿过细胞膜进入细胞内,亲水性的PEG也被引入额外包被在纳米粒子表面提高亲水性。
因此,利用壳聚糖与PLGA的共价连接,通过PLGA纳米粒子表面壳聚糖的引入来减轻异物排斥反应,降低巨噬细胞对疏水纳米粒子的吞噬成为提高载药纳米靶向性的新途径。本文以人乳腺癌细胞MCF-7作为培养细胞,阿霉素作为抗癌药物,来研究载药壳聚糖PLGA纳米载体对肿瘤细胞生长的抑制作用,同时对比传统方法合成的PLGA纳米载体和壳聚糖表面修饰PLGA得到的纳米载体(C-NPs),对自组装的壳聚糖PLGA纳米载体(G-NPs)作为新型纳米抗癌药物载体用于肿瘤细胞抑制的定性和定量研究,通过荧光观察肿瘤细胞对纳米粒的摄取情况,进而评价G-NPs作为新型纳米载体在肿瘤治疗方面的应用。
1 材料
1.1 药品与试剂
壳聚糖(chitosan,CHS,26kDa,DD=90%),购自山东恒台县金湖甲壳制品有限公司;端羧基聚乳酸羟基乙酸(poly(lactic-co-glycolic acid,PLGA,50/50,Mv=5 000,购自山东省医疗器械研究所;异硫氰酸荧光素(Fluorescein isothiocyanate,FITC)购自Sigma公司;冰醋酸、乙醇、盐酸、氢氧化钠等其他试剂均为分析纯;人乳腺癌细胞MCF-7由青岛大学医学院附属医院中心实验室提供。
1.2 主要仪器设备
VCX 105型超声仪(美国Uibra公司);HealForce 90二氧化碳培养箱(河南兄弟仪器设备有限公司);UV-1601紫外分光光度计(日本Shimadzu公司);Biotek Synergy2多功能荧光酶标仪(美国Bioteck公司);XDS-1倒置显微镜(南京米厘特精密仪器有限公司);BX51荧光显微镜(日本Olympus公司)。
2 方法
2.1 空白及载药纳米载体的制备
空白PLGA纳米粒子的制备参照 Wang等[6]的方法,具体步骤如下:首先,称取100mg PLGA溶解于5mL二氯甲烷中,然后逐滴加入到含0.5%(w/v)PVA的去离子水中,磁力搅拌器持续搅拌形成初乳,然后用超声均质机以800r/min超声10min,利用旋转蒸发仪旋蒸掉有机溶剂得到PLGA纳米分散体系,以15 000r/min离心10min,去离子水洗涤3次,重复离心得到的产物冻干备用。
表面修饰的壳聚糖-PLGA纳米载体(C-NPs)的制备:称取100mg制得的PLGA纳米粒子分散于5mL MES缓冲液中(pH=5.0),加入一定量的1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC)和N-羟基琥珀酰亚胺(NHS)活化反应一定时间,然后将50mL配置好的0.2%(w/v)壳聚糖溶液加入以上反应体系,室温下搅拌反应48h,15 000r/min离心10 min后冷冻干燥得到产物C-NPs。在乳化前,将一定量阿霉素预溶于油相二氯甲烷中,即得载阿霉素的PLGA纳米粒子和C-NPs。
自组装的壳聚糖-PLGA纳米载体(G-NPs)的制备:称取100mg PLGA溶解于5mL的二氯甲烷中,将以上溶液滴加入含一定量EDC和NHS的5mL MES缓冲液中(pH=5.0)冰浴下活化1.5h,然后将50mL配置好的0.2% (w/v)壳聚糖溶液加入到以上反应体系,室温下搅拌反应48h,旋蒸掉有机溶剂,过滤后以去离子水透析48h去除过量反应底物,冷冻干燥制得聚合物。称取一定量制备得到的聚合物分散于去离子水中,在冰浴条件下以超声仪超声3min(超声间隔,超3s停2s,功率30%),得到的样品溶液以0.8μm滤膜过滤后冷冻干燥,即得G-NPs。
载药G-NPs的制备参照Hyung等[7]的方法,将一定量阿霉素分散于PBS中(pH=7.4),加入制备得到的聚合物,超声制备得到包载DOX的G-NPs,实验过程避光,制得样品冻干保存备用。
2.2 FITC标记的空白纳米粒子FITC-C-NPs和FITCG-NPs的制备
分别称取40mg精制的C-NPs和G-NPs超声分散于20mL去离子水中,用1mol/L的NaOH调节pH至7.5。称取FITC 4mg溶于4mL无水甲醇中,逐滴加入到含CP和GP的去离子水中,室温避光反应6h后,向反应体系中加入48mL的甲醇/氨水(v/v,7∶3)的混合液,8 000×g离心10min,弃上清液,所得沉淀用甲醇洗涤至滤液在495nm下无荧光吸收为止。冷冻干燥后得黄色目标产物。
2.3 MCF-7细胞对纳米粒摄取的定量测定
MCF-7细胞对荧光纳米粒摄取的定量测定参照Khin等[8]的方法:取指数生长期 MCF-7细胞,用胰酶消化后接种于96孔细胞培养板上(100μL/孔,接种密度为3×104个/mL),在37℃、5%CO2、95%湿度的二氧化碳培养箱中培养12h至细胞贴壁生长成单层,吸出原培养液,用5mL D-Hank’s缓冲液于培养箱中平衡培养30min,吸除D-Hank’s后,加入不同浓度的以D-Hank’s配置的荧光纳米悬液(FITC-C-NPs 和FITC-G-NPs),纳米悬液浓度分别为:25,50,100,200,400和800μg/mL,于二氧化碳培养箱中分别培养1,2和4h后,将荧光纳米粒悬液吸出,以PBS缓冲液(pH=7.4)轻轻漂洗3次除去游离纳米粒,每孔加入100μL 0.5%的Triton X-100细胞裂解液裂解细胞30min,最后于荧光酶标仪检测分离得到的细胞裂解液的荧光强度(λex=485nm,λem=528nm)。其中未处理的空白细胞作为对照,参照FITC标准曲线计算细胞中的FITC含量,最后根据公式(1)计算 MCF-7细胞对2种不同浓度的荧光纳米粒子的摄取效率(Cellular Uptake Efficiency):
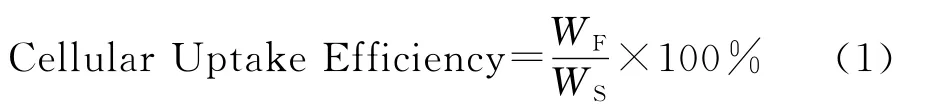
其中:WF为不同浓度荧光纳米粒子处理得到的细胞裂解液中FITC的含量;WS为不同浓度荧光纳米粒子中FITC的总含量。
2.4 细胞吞噬荧光纳米粒的显微镜观察
MCF-7细胞对FITC-C-NPs和 FITC-G-NPs两种纳米粒的摄取参照Zhang等[9]的方法,取传代至指数生长期的MCF-7细胞用0.25%胰酶细胞消化后,分别取1mL/孔接种到6孔培养板中(接种密度为3×104个/mL),在37℃、5%CO2、95%湿度的二氧化碳培养箱中培养12h至细胞贴壁生长成单层,每孔加入一定浓度的 FITC-C-NPs和 FITC-G-NPs纳米粒溶液,分别于CO2培养箱中继续培养1和4h,培养完成后,用pH=7.4的PBS缓冲液小心漂洗细胞3次,于荧光显微镜下进行观察并拍照。
2.5 载药纳米粒对肿瘤细胞生长抑制的定量测定
DOX,DOX-PLGA NPs,DOX-C-NPs和 DOX-GNPs对肿瘤细胞生长抑制的定量测定,作者采取MTT法[10]:取指数生长期 MCF-7细胞,用1mL 0.25%胰酶消化后,加入10mL新鲜培养液(DMEM高糖培养基,10%胎牛血清),取100μL/孔接种于96孔细胞培养板上(接种密度3×104个/mL),在37℃、5%CO2、95%湿度的二氧化碳恒温培养箱中孵育过夜,至MCF-7细胞贴壁生长成单层后,吸出原培养液,每孔加入不同药物浓度的3种载药纳米粒溶液(DOX-PLGA NPs、DOX-C-NPs和DOX-G-NPs)和游离阿霉素(200μL/孔,每个浓度设5个平行),用含10%胎牛血清(FBS)的DMEM培养液作为阴性对照,继续培养一定时间。每孔加入20μL 5mg/mL的MTT溶液反应,继续孵育4 h后弃去上清液,每孔加入150μL DMSO,使结晶物充分融解,终止反应,恒温振荡反应10min后,在酶标仪490nm处测定培养板各组样品的吸光度值,按公式(2)计算载药纳米粒对MCF-7细胞生长的相对抑制率(Inhibitory rate of control,IR%):
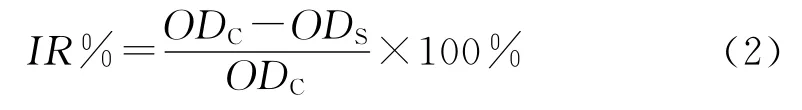
其中:ODC为阴性对照组的吸光度值;ODS为样品处理组的吸光度值。
3 结果与讨论
3.1 3种纳米载体的合成
经乳化方法制备的空白PLGA NPs为圆球形(见图1);C-NPs形成的是以PLGA纳米球为核心的表面包被壳聚糖的结构;G-NPs则是通过链与链间共价接枝聚合,在超声作用下自组装而成的结构均一的纳米粒子。显然对比C-NPs与G-NPs,两者虽然都是壳聚糖和PLGA通过酰胺键共价连接而成,但由于合成路线和成球方法的差别,两者具有不同的纳米结构,而作为纳米载体的纳米结构的影响,则可通过结构的稳定性,药物包载和释放,细胞吞噬效率等研究来分析。
3.2 MCF-7细胞对荧光纳米粒摄取的定量测定
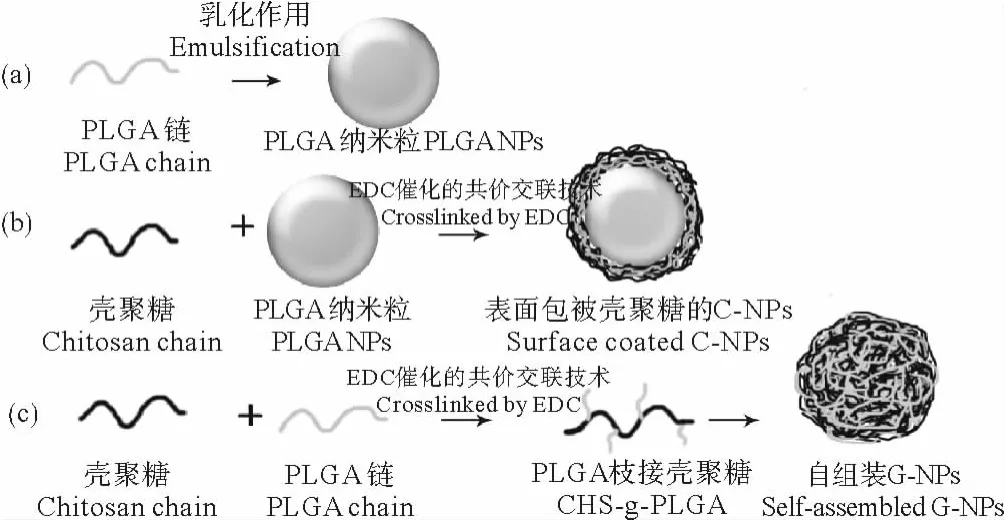
图1 3种纳米载体的合成Fig.1 Schematic representation of three differently structured nanoparticles
制备得到的不同浓度C-NPs和G-NPs 2种荧光标记纳米粒子在MCF-7细胞中孵育1h后,细胞对纳米粒的摄取情况见图2,随着纳米粒子浓度从25μg/mL升高到400μg/mL,MCF-7细胞对2种荧光纳米粒的摄取效率逐渐增加,呈一定的浓度依赖性,C-NPs和GNPs 2种纳米粒在细胞孵育1h后的荧光摄取效率没有明显差异;而当纳米粒子的浓度增加到800μg/mL时,细胞对荧光纳米粒的摄取效率开始出现下降,CNPs和G-NPs分别从400μg/mL浓度时的6.74%和6.92%,下降到800μg/mL浓度时的5.36%和5.24%。
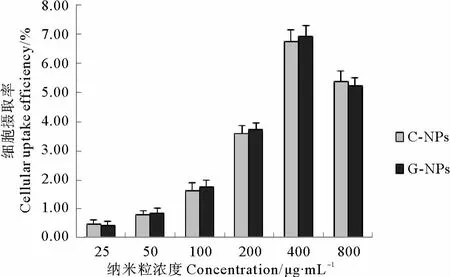
图2 孵育1h后MCF-7细胞对2种不同浓度纳米粒子的细胞摄取效率Fig.2 Cellular uptake efficiency of different concentration nanoparticles by MCF-7cells after 1hincubation
研究表明,细胞对纳米粒子的摄取主要受纳米粒子的浓度、表面电荷、粒径、形状及表面修饰基团理化性质的 影 响[11-12],MTT实验已经表明,C-NPs和 GNPs由于壳聚糖的修饰作用,其相对细胞毒性高于PLGA纳米粒子,表明壳聚糖的修饰增加了纳米粒子对细胞的黏附作用。壳聚糖主链上活性氨基基团质子化后,通过与细胞膜表面负电蛋白的非特异性静电吸附而促进粒子与细胞的黏附,当纳米粒子到达细胞膜后,再通过细胞膜上亲水基团或疏水基团与纳米粒子之间的相互作用,细胞膜内陷包裹纳米粒子进入细胞内。Guan等[13]和 Pamujula等[14]报道壳聚糖亲水性和氨基阳离子有助于细胞对纳米粒子的黏附,这种黏附作用随着纳米粒子浓度的增加而提高。纳米粒子的表面亲水性降低了巨噬细胞吞噬系统对纳米粒子的吞噬,进一步增加细胞对纳米粒子的有效黏附和摄取效率[15]。
因此低浓度条件下(25~400μg/mL),纳米粒子表面的亲水性和表面电荷促进了MCF-7细胞对2种纳米粒子的黏附作用,细胞对2种纳米粒子的摄取都呈现出浓度依赖性,随浓度的升高而增大,而当纳米粒浓度增大至800μg/mL时,细胞对纳米粒的摄取量逐渐趋近饱和,导致了细胞相对摄取效率下降。
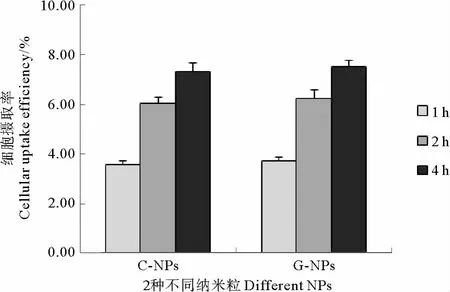
图3 孵育不同时间后MCF-7细胞对浓度为200μg/mL的2种纳米粒的细胞摄取效率Fig.3 Cellular uptake efficiency of different nanoparticles by MCF-7cells incubated for different times with a particle concentration of 200μg/mL
为了进一步研究MCF-7细胞对荧光纳米粒子摄取随时间变化的关系,以浓度为200μg/mL的C-NPs和G-NPs分别孵育细胞1、2、4h后,测定 MCF-7细胞对纳米粒子的摄取效率(见图3)。MCF-7细胞对CNPs和G-NPs的摄取也呈现出时间依赖性,随着孵育时间的延长,细胞摄取效率逐渐增加,C-NPs从1h的3.59%升高到了4h后的7.33%;G-NPs从1h时的3.71%升高到了4h后的7.51%。
通过测定MCF-7细胞对C-NPs和G-NPs摄取效率随浓度和时间变化的关系,说明自组装合成的GNPs与传统方法制备得到C-NPs相比,由于2种纳米粒子的表面结构和性质存在差异,在表面亲水性壳聚糖和活性氨基的作用下,其细胞吞噬效率在4h内未表现出明显差别,MCF-7细胞对2种纳米粒子的摄取,在浓度为25~400μg/mL,孵育1~4h的条件下,摄取效率具有浓度和时间依赖性。
3.3 细胞摄取荧光纳米粒的显微镜观察
MCF-7细胞对2种空白荧光标记纳米粒子摄取情况的荧光显微镜照片见图4,A/B为FITC-C-NPs组;C/D为FITC-G-NPs组;A/C为显微镜光源下荧光未激发的细胞形态照片、B/D为荧光蓝色通道激发FITC后的显色照片;细胞孵育荧光纳米粒1h后,即可看到2种FITC标记的纳米粒子已经进入细胞内(呈绿色荧光标记),细胞对2种纳米粒子的摄取情况无明显差异,与定量测定的结果相一致。

图4 荧光倒置显微镜观察细胞对纳米粒子摄取Fig.4 Fluorescence microscopic images of different nanoparticles with MCF-7cells
为了观察C-NPs和G-NPs 2种纳米粒子细胞摄取孵育时间的变化,同时观察包载药物阿霉素后,MCF-7细胞对纳米药物载体的摄取分布情况,作者制备了FITC标记的包载阿霉素的2种纳米粒子:FITC-DOXC-NPs和FITC-DOX-G-NPs,在 MCF-7细胞孵育4h后,通过荧光显微镜观察纳米粒子和药物在细胞内的分布情况,见图5,A/B为 FITC-DOX-C-NPs组;C/D为FITC-DOX-GNPs组;A/C 为荧光绿色通道激发DOX后的显色照片(红色)、B/D为荧光蓝色通道激发FITC后的显色照片(绿色);可以明显看出包载药物的纳米粒子已经通过细胞内吞作用进入到了细胞膜内,DOX和FITC呈现相同的标记位置,证明荧光标记的载药纳米粒子成功进入到了细胞内。FITC-DOX-CNPs和 FITC-DOX-G-NPs 2种纳米粒子在孵育4h后,在细胞内的数量明显增加(对比图4),结合定量测定的结果,进一步说明MCF-7细胞对2种纳米粒子的摄取具有时间依赖性。而且细胞在孵育1和4h后,细胞对2种纳米粒子的吞噬情况无明显差异。

图5 孵育4h后MCF-7细胞对2种载药纳米粒子的细胞摄取Fig.5 Fluorescence microscopic images of different nanoparticles with MCF-7cells incubated for 4h
3.4 载药纳米粒对肿瘤细胞生长抑制的定量测定
通过MCF-7细胞对空白荧光纳米粒摄取的定量测定和观察,证明了经壳聚糖修饰的2种纳米粒子CNPs和G-NPs可以有效的被细胞吞噬,然而药物包载后,经壳聚糖修饰的载药纳米粒子药物释放效果的评价,则需要进一步通过肿瘤细胞生长的抑制来进行测定。
图6为3种载药纳米粒子对比游离DOX在细胞孵育12h后,细胞生长抑制率随药物浓度变化的情况。PLGA、C-NPs、G-NPs和游离药物对 MCF-7细胞生长的抑制率都随着药物浓度的增加而增加,在低浓度条件下(1~4μg/mL),3种载药纳米粒子的细胞生长抑制率基本都要高于游离药物组,而当药物浓度从8μg/mL上升到16μg/mL时,游离药物的细胞生长抑制率快速升高(16μg/mL,58.77%),超过了3种载药纳米粒子(16μg/mL PLGA 33.07%;C-NPs 39.66%;GNPs 40.05%),说明在较低药物浓度下,通过纳米药物载体的靶向性和细胞吞噬增加了细胞对纳米药物载体的摄取,而纳米载体对P-gp主导的MDR效应的削弱进一步提高了细胞内药物的有效浓度,而游离阿霉素在较低浓度下,无法形成较高的渗透压,而导致药物扩散作用相对较弱,而且药泵P-gp的作用和溶酶体的吞噬溶解,进一步降低了游离药物在细胞内的积聚;而随着药物浓度的升高(16μg/mL),游离药物的扩散作用增强,细胞内游离阿霉素的浓度迅速增加,而纳米载体药物的缓释性使得在12h的孵育条件下,高浓度的游离药物明显高于包载药物的纳米载体对细胞生长的抑制作用。
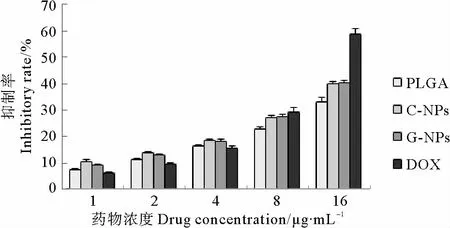
图6 孵育12h载药纳米粒对MCF-7细胞生长的抑制率Fig.6 Inhibitory rate of drug-loaded nanoparticles to MCF-7cells incubated for 12h
同时在12h孵育条件下,3种纳米药物载体中,CNPs和G-NPs在不同的药物浓度下均表现出比PLGA NPs高的细胞生长抑制率,这主要是由于C-NPs和GNPs纳米载体表面壳聚糖亲水性基团和正电性氨基的存在,使得这2种纳米药物载体的靶向性和细胞黏附效率明显增加,从而表现出相对于PLGA纳米载体更好的细胞生长抑制效果。而在12h孵育条件下,未观察到C-NPs和G-NPs的细胞生长抑制差异。
4 结语
以人乳腺癌细胞MCF-7为模型细胞对荧光标记纳米载体的摄取和生长抑制情况进行了研究,壳聚糖修饰的2种纳米粒子C-NPs和G-NPs表现出良好的细胞靶向性,通过荧光显微镜观察,均能有效的被MCF-7细胞摄取,而2种壳聚糖修饰的纳米载体的摄取效率无显著差异。细胞生长抑制的定量测定表明,由于壳聚糖的靶向作用,载药后C-NPs和G-NPs在低药物浓度1~4μg/mL时,具有优于游离阿霉素和载药PLGA纳米载体的肿瘤细胞生长抑制率,另外载药纳米载体对MCF-7细胞生长的抑制具有一定的时间和浓度依赖性。结果表明,G-NPs包载疏水抗癌药物后,有潜力成为抑制肿瘤细胞生长的新型纳米载体。
[1]Yang X H,Sladek T L,Liu X S,et al.Reconstitution of caspase 3 sensitizes MCF-7breast cancer cells to doxorubicin-and etoposideinduced apoptosis[J].Cancer Research,2001,61(1):348-354.
[2]Bae Y,Diezi T A,Zhao A,et al.Mixed polymeric micelles for combination cancer chemotherapy through the concurrent delivery of multiple chemotherapeutic agents[J].J Control Release,2007,122(3):324-330.
[3]De S,Miller D W,Robinson D H.Effect of particle size of nanospheres and microspheres on the cellular-association and cytotoxicity of paclitaxel in 4T1cells[J].Pharm Res,2005,22(5):766-775.
[4]Gref R,Domb A,Quellec P,et al.The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres[J].Adv Drug Delivery Rev,2005,16(2-3):215-233.
[5]Hu F Q,Meng P,Dai Y Q,et al.PEGylated chitosan-based polymer micelle as an intracellular delivery carrier for anti-tumor targeting therapy[J].Eur J Pharm Biopharm,2008,70(3):749-757.
[6]Wang Z H,Wang Z Y,Sun C S,et al.Trimethylated chitosanconjugated PLGA nanoparticles for the delivery of drugs to the brain[J].Biomaterials,2010,31(5):908-915.
[7]Hyung P J,Kwon S,Lee M,et al.Self-assembled nanoparticles based on glycol chitosan bearing hydrophobic moieties as carriers for doxorubicin:In vivo biodistribution and anti-tumor activity[J].Biomaterials,2006,27(1):119-126.
[8]Khin Y W,Feng S S.Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs[J].Biomaterials,2005,26(15):2713-2722.
[9]Zhang L K,Hou S X,Mao S J,et al.Uptake of folate-conjugated albumin nanoparticles to the SKOV3cells[J].Int J Pharm,2004,287(1-2):155-162.
[10]Mosmann T.Rapid colorimetric assay for cellular growth and survival:application to proliferation and cytotoxicity assays[J].J Immunol Methods,1983,65(1-2):55-63.
[11]Rejman J,Oberle V,Zuhorn I S,et al.Size-dependent internalization of particles via the pathways of clathrin-and caveolae-mediated endocytosis[J].Biochem J,2004,377(1):159-169.
[12]Carlson C,Hussain S M,Schrand A M,et al.Unique cellular interaction of silver nanoparticles:size-dependent generation of reactive oxygen species[J].J Phys Chem B,2008,112(43):13608-13619.
[13]Guan X P,Quan D P,Liao K,et al.Preparation and characterization of cationic chitosan-modified poly(d,l-lactide-coglycolide)copolymer nanospheres as DNA carriers[J].J Biomater Appl,2008,22(4):353-371.
[14]Pamujula S,Graves R A,Moiseyev R,et al.Preparation of polylactide-co-glycolide and chitosan hybrid microcapsules of amifostine using coaxial ultrasonic atomizer with solvent evaporation[J].J Pharm Pharmacol,2008,60(3):283-289.
[15]Owens D E,Peppas N A.Opsonization,biodistribution,and pharmacokinetics of polymeric nanoparticles[J].Int J Pharm,2006,307(1):93-102.
