通过CRISPR/Cas9系统敲除人源PDE10A基因
2014-11-27梁振伟饶书权
梁振伟,饶书权,沈 岩,许 琪
(中国医学科学院 基础医学研究所 医学分子生物学国家重点实验室,北京 100005)
通过CRISPR/Cas9系统敲除人源PDE10A基因
梁振伟,饶书权,沈 岩,许 琪*
(中国医学科学院 基础医学研究所 医学分子生物学国家重点实验室,北京 100005)
目的建立敲除基因组中PDE10A基因的CRISPR/Cas9系统。方法设计3个长20bp的sgRNA,分别靶向PDE10A的exon 6、exon 7和exon 11。化学合成sgRNA寡核苷酸序列,并克隆进PX330质粒中。将克隆正确的质粒PX330-sgRNA转染至HEK293T细胞中,提取基因组DNA并对敲除位点附近的DNA片段进行PCR扩增,再通过SURVEYOR分析和一代测序对敲除效率进行检测。最后采用有限稀释法挑选稳定敲除PDE10A的HEK 293T细胞株。结果目的sgRNA 寡核苷酸双链成功插入PX330质粒中且序列正确;靶向Exon 7的sgRNA可成功敲除PDE10A,且敲除效率高达31.4%;稳定敲除PDE10A的细胞株筛选成功,可导致2 bp 缺失。结论PDE10A基因CRISPR/Cas9敲除系统构建成功。
PDE10A;CRISPR/Cas9;稳定细胞株
PDE10A基因编码一种属于环核苷酸磷酸二酯酶家族的蛋白[1],通过水解cAMP和cGMP的5′磷酸基团,调节细胞内环核苷酸的浓度,从而在细胞信号传导中起重要作用。PDE10A主要分布于新纹状体中具有多巴胺神经元活性的中间棘神经元[2],在脑内其他组织如海马、皮质及小脑等也有少量表达[3]。PDE10A表达量异常升高与多种涉及基底神经节神经传递的神经精神系统疾病的发生有关,如精神分裂症、强迫症和亨廷顿氏舞蹈病等。当使用PDE10A的抑制因子时,可以在小鼠和恒河猴中产生类似抗精神病药物的效应,改善动物的认知功能[4]。本研究利用CRISPR/Cas9建立PDE10A基因靶向敲除系统,为研究PDE10A基因的功能和基因敲除干细胞或小鼠提供一部分研究依据。
1 材料与方法
1.1 材料
HEK 293T细胞(中国医学科学院基础医学研究所细胞中心),PX330质粒,DH5a感受态(Transgene公司),限制性内切酶BbsⅠ和T4连接酶(NEB公司),质粒小提试剂盒,2×PCR mix(天根生化科技有限公司),DMEM高糖培养基(Thermo公司),NeoFect DNA转染试剂(零客创智有限公司),琼脂糖胶回收试剂盒(康为世纪公司),FlexiGene Kit(Qiagen公司),SURVEYOR分析试剂盒(Transgenomic公司)。
1.2 方法
1.2.1 sgRNA靶点选择及其寡核苷酸链合成:应用http://crispr.mit.edu/网站设计PDE10Aguide RNA。设计标准如下:1)长20 nt的寡核苷酸sgRNA核心序列按G(N)20NGG序列进行设计;2)正义链模板的5′端添加CACC,与BbsⅠ酶切后形成的黏性末端互补;反义链模板的5′端添加AAAC,与BbsⅠ酶切后形成的黏性末端互补。寡核苷酸序列(表1)。设计CRISPR 鉴定引物:1)PDE10A-Exon 7:F:5′-GATA GAAATAAGAATCATCAC-3′,R:5′-TCTCCAAAGTC CTTCTACTAC-3′,切割后,上游191 bp,下游328 bp;2)PDE10A-Exon 6:F:5′-GTGAAGTGGAAATGTGA GATAG-3′,R:5′-TGTTAGTGGGTAAAG GGAA-3′,切割后,上游190 bp,下游325 bp; 3)PDE10A-Exon 11:F:5′-CTAATCAGAATGAGTGGTGGTCG-3′,R:5′-CA CAGAAAAATGGGTAACGGA-3′,切割后,上游474 bp,下游177 bp。所有寡核苷酸链和引物均由Invitrogen公司合成。
1.2.2 PX330-sgRNA载体构建:将sgRNA寡核苷酸单链退火形成双链:取等量的上游链和下游链混合(终浓度为50 μmol/L)。程序如下:95 ℃,5 min;94 ℃,1 min,-1 ℃ /循环,22个循环;72 ℃,30 min;71 ℃,1 min,-1 ℃/循环,46个循环;4 ℃保持。将sgRNA寡核苷酸双链稀释至0.5 μmol/L。用BbsⅠ内切酶线性化PX330质粒,切胶回收后稀释至50 g/L。连接体系:线性化PX330质粒,50 ng;0.5 μmol/L sgRNA寡核苷酸双链,1 μL;10×T4 DNA连接酶缓冲液,1.5 μL;牛血清白蛋白(10 g/L):0.5 μL;加水至15 μL,16 ℃连接过夜。转化DH5α感受态,挑取单克隆,小提质粒并测序验证(由诺赛基因公司完成)。
1.2.3 细胞培养和细胞转染:HEK293T细胞培养条件:高糖-DMEM培养基(含10%胎牛血清),5% CO2,37 ℃恒温培养。
转染前24 h,将HEK293T细胞以5×105/孔接种至6孔板中培养,转染时细胞汇合度达到60 %~70 %。转染前1 h更换新鲜培养基,采用NeoFect转染试剂进行细胞转染。等量pEGFP-N1质粒作为阴性对照。
1.2.4 细胞基因组DNA提取:转染48 h后,将各组细胞消化收集,用磷酸盐缓冲液洗涤3次,采用FlexiGene Kit试剂盒提取基因组DNA[5]。
1.2.5 PCR,SURVEYOR分析以及一代测序鉴定:PCR条件如下:20 μL PCR体系:2×PCR Mix:10 μL;ddH2O:8 μL;基因组DNA:1 μL(50 ng);10 μmol/L Primer(上游和下游)各0.5 μL,共20 μL。PCR程序为:94 ℃ 5 min,35个循环(94 ℃ 30 s,50~60 ℃ 30 s,72 ℃ 1 min),4 ℃保持。取2 μL产物进行琼脂糖凝胶电泳(浓度1%)检测。对sgRNA靶点附近的基因组DNA扩增后进行正向测序,若sgRNA位点以后的区域出现低矮的套峰,说明基因敲除成功。
SURVEYOR分析按如下步骤进行:用1 %琼脂糖凝胶对PCR产物进行切胶纯化。将回收产物稀释至50 g/L,取20 μL回收产物按照SURVEYOR分析试剂盒操作步骤进行[6]检测,用2 %琼脂糖凝胶进行分析。随后,进行吸光度分析,计算公式为:indel (%)=100×[1-(1-fcut)1/2],fcut=(b+c)/(a+b+c),其中a表示未被切割条带的吸光度值,b和c分别表示切割产生新条带的吸光度值,indel为插入缺失率,fcut为切割比率。
1.2.6 筛选PDE10A稳定敲除细胞株:将PX330-sgRNA质粒转染HEK 293T细胞48 h后,采用有限稀释法,将单细胞种至96孔板中。操作步骤如下:胰蛋白酶消化细胞并计数,采用梯度稀释法稀释至每100 μL培养基0.5个细胞,按每孔100 μL细胞稀释液加至96孔板中。待细胞生长至96孔板底1/2时,取少量细胞提取DNA,经PCR扩增后测序,测序结果与原基因组DNA进行对比,检测是否靶向PDE10A成功[7]。
2 结果
2.1 sgRNA靶点以及寡核苷酸序列设计
PX330质粒信息(图1A);以Exon 7 sgRNA为例,其插入位置和序列信息(图1B)。sgRNA靶点以及寡核苷酸序列(表1)。

A.the structure of plasmid PX330, which has twoBbsⅠ restriction sites in the target sites; B.the genome locus of Exon 7 sgRNA

图1 PX330载体构建示意图Fig 1 The establishment of plasmid PX330
2.2 PX330-sgRNA质粒的测序结果
插入序列的位置、方向及序列与预期相符,证明质粒构建完全正确(图2)。
2.3SURVEYOR分析对sgRNA靶向敲除效果进行检测
只有Exon 7 sgRNA靶向敲除成功(图3)。
2.4 利用一代测序对靶向敲除效果进一步检测
Exon 7 sgRNA 靶点附近的基因组DNA的一代测序(图4)。
2.5 建立稳定敲除PDE10A的293T细胞株
单克隆细胞测序后与原基因组DNA进行对比结果(图5)。
3 讨论
传统基因敲除,主要是应用同源重组原理通过插入突变和基因靶向技术使目的基因的功能丧失[8]。而CRISPR/Cas系统,利用与目的基因具有同源性的sgRNA能够特异性结合基因组DNA上,形成杂合双链,而特定的核酸内切酶Cas9能够对该杂合双链进行切割造成DNA的双链断裂(double-strand breaks, DSB)。DSB诱导产生位点特异性的核酸酶,通过错误倾向的非同源末端连接 (non-homologous end joining, NHEJ)对切断的双链进行修复,这种修复将在断裂位点产生插入或者缺失。所以与特定位点同源的sgRNA以及Cas9是进行基因敲除不可缺少的两个元件。该系统具有快速、可靠、特异性高以及敲除效率高等特点[9]。

The nucleotides in italic represented sgRNA sequences图2 插入DNA序列测序图Fig 2 Sequences of the inserts
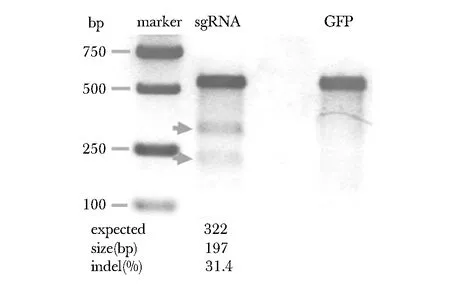
The marker is D2000 and the GFP was used as negative control图3 SURVEYOR 检测Exon 7 sgRNA的靶向敲除效果Fig 3 SURVEYOR assay of Exon 7 sgRNA targeted knockout effect
本研究构建的PX330-PDE10A-Exon 7-sgRNA质粒携带有化脓性链球菌型Ⅱ型CRISPR/Cas系统的Cas9核酸内切酶基因,在转染进细胞后可以表达该核酸内切酶。通过SURVEYOR分析试剂盒进行产物多态性分析和特定位点的切割效率检测,发现PX330-PDE10A-Exon 7-sgRNA的转染在PDE10A基因目的位点产生切割的效率高达31.4%。通过一代测序,可以发现在特定位点存在大量的套峰结构,这进一步证实了SURVEYOR分析的结果。在28个细胞亚克隆中,有3个克隆在目的片段中出现了碱基的缺失突变,其中3 bp和6 bp缺失对PDE10A编码的蛋白序列影响较小,选取缺失2 bp的细胞株进行扩大培养,建立稳定敲除PDE10A的细胞株。这进一步证明该系统对PDE10A基因的敲除是有效的。
由于PDE10A蛋白主要调节细胞中cAMP的浓度,而cAMP是细胞内重要的第二信使分子,在许多细胞信号通路中存在影响。因此,PDE10A基因在多种神经精神系统疾病的病理学发生途径中具有重要作用[10]。本研究构建PDE10A基因CRISPR/Cas9敲除系统,将为后续构建PDE10A敲除干细胞和敲除小鼠提供实验依据。
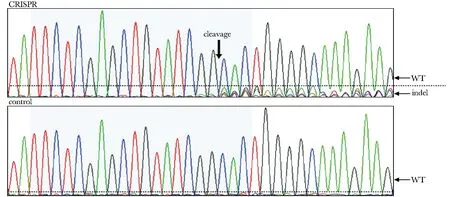
As is noted that the cleavage site was located just 3bp before the PAM motif图4 一代测序验证Exon 7 sgRNA靶向敲除效果Fig 4 The sequences of the Exon 7 sgRNA target site

图5 3个单克隆细胞在目的sgRNA位点的缺失突变Fig 5 The deleted mutation of the three monoclonal cells at the sgRNA sites
[1] Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents[J]. Pharmacol Ther, 2006, 109: 366-398.
[2] Menniti FS, Chappie TA, Humphrey JM,etal. Phosphodiesterase 10A inhibitors: a novel approach to the treatment of the symptoms of schizophrenia[J]. Curr Opin Investig Drugs,2007, 8: 54-59.
[3] Kilburn JP, Kehler J, Langgard M,etal. N-Methylanilide and N-methylbenzamide derivatives as phosphodiesterase 10A (PDE10A) inhibitors[J]. Bioorg Med Chem, 2013, 21: 6053-6062.
[4] Cutshall NS, Onrust R, Rohde A,etal. Novel 2-methoxyacylhydrazones as potent, selective PDE10A inhibitors with activity in animal models of schizophrenia[J]. Bioorg Med Chem Lett, 2012, 22: 5595-5599.
[5] Martin-Nunez GM, Gomez-Zumaquero JM, Soriguer F,etal. High resolution melting curve analysis of DNA samples isolated by different DNA extraction methods[J]. Clin Chim Acta, 2012, 413: 331-333.
[6] Gennequin B, Otte DM, Zimmer A. CRISPR/Cas-induced double-strand breaks boost the frequency of gene replacements for humanizing the mouse Cnr2 gene[J]. Biochem Biophys Res Commun, 2013, 441: 815-819.
[7] Yang H, Wang H, Shivalila CS,etal. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering[J]. Cell,2013, 154: 1370-1379.
[8] Lamotte L, Jackerott M, Bucchini D,etal. Knock-in of diphteria toxin A chain gene at Ins2 locus: effects on islet development and localization of Ins2 expression in the brain[J]. Transgenic Res, 2004, 13: 463-473.
[9] Fu Y, Foden JA, Khayter C,etal. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells[J]. Nat Biotechnol,2013, 31: 822-826.
[10] Grauer SM, Pulito VL, Navarra RL,etal. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia[J]. J Pharmacol Exp Ther,2009, 331: 574-590.
Knocking out humanPDE10Agene by CRISPR/Cas9 system
LIANG Zhen-wei, RAO Shu-quan, SHEN Yan, XU Qi*
(State Key Laboratory of Medical Molecular Biology,Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Beijing 100005, China)
ObjectiveTo construct the CRISPR/Cas9 system constructed by knocking out thePDE10Agene.MethodsThree 20 bp sgRNAs targeting exon 6, exon 7 and exon 11 ofPDE10Awere designed, chemically synthesized, and then inserted into linearized plasmid, PX330. Next, the correct PX330-sgRNA plasmids were transfected into HEK293T cells after verification by Sanger sequencing. The targeting efficiency was detected by SURVEYOR assay and the nicked site was further detected by Sanger sequencing. Finally, the stablePDE10Aknockout 293T cell line was selected by the limiting dilution method.ResultsThe target nucleotide sequences were successfully inserted into the expected sites of vector and sequences were correct. The targeted exon 7 sgRNA can successfully knockPDE10A, and the targeting efficiency was up to 31.4 %. Stable knockoutPDE10Acell line was selected successfully which harbored 2 bp deletion.ConclusionsThe CRISPR/Cas9 system constructed by knocking out thePDE10Agene is successfully constructed.
PDE10A; CRISPR/Cas9 system; Stable cell lines
2013-12-19
2014-01-07
国家自然科学基金(31222031);中央高校基本科研业务费专项资金(2012S05);协和青年科研基金(2012J09);新世纪优秀人才支持计划(NCET-12-0071)
*通信作者(correspondingauthor): xuqi@pumc.edu.cn
1001-6325(2014)04-0439-05
研究论文
R 34
A
