用密度泛函理论研究两种金属镍席夫碱配合物的电子结构和光谱性质
2014-11-27李洁琼李永红
李洁琼,李永红
(河南大学 化学化工学院,河南 开封 475004)
近年来,席夫碱作为一类重要的生物配体引起了科学界的广泛关注.这类配体不仅含有N、S、O原子等多个配位点而且具有多种配位形式,可以与金属形成结构新颖的多位点配合物[1-2].席夫碱金属配合物不但具有独特的抗病毒、抗癌和抑制细菌生长等生物和药用价值,而且还是一种广为使用的催化剂,已经成为药物化学、工业催化和农药等方向的研究宠儿[3].席夫碱及其金属配合物的各种生物及催化活性取决于席夫碱的结构和不同的金属离子,了解不同配合物的结构与性质的关系对合成新功能、新用途的席夫碱金属配合物起着重要的作用.
ASADI等人[4]用金属镍分别与两种席夫碱配体配位合成了金属配合物1(C27H16N2Br2O3Ni)和2(C27H16N4O7Ni).但是,实验上对于这些配合物的几何构型参数和电子性质没有给出详尽的报道,且实验上虽给出了吸收峰的位置,但没有对它们吸收光谱曲线及跃迁性质做出进一步研究.本文作者采用密度泛函理论在B3LYP方法下以6-31+G(d)-LANL2DZ为基组研究了这两个配合物的几何结构,前线分子轨道能量及其分布情况,然后基于优化的几何结构使用含时密度泛函理论计算了它们在N,N-二甲基甲酰胺溶液中的电子吸收光谱.
1 计算方法
采用密度泛函理论(DFT)的B3LYP[5-6]方法,对配合物1和2的几何构型进行全优化,并且对所有的优化构型进行了频率分析.对于C,H,O,N和Br原子,采用6-31+G(d)基组[7-8];对于Ni,采用LANL2DZ赝势基组[9-10].对于基态构型,所有计算得到的频率都是正的,说明优化得到的结构均为稳定构型.在此基础上,应用含时密度泛函理论(TD-DFT),在TD-B3LYP/6-31+G(d)-LANL2DZ水平下分别计算了这两种配合物在N,N-二甲基甲酰胺溶液中的电子吸收光谱.溶剂化效应采用极化连续介质模型(PCM)[11-12].所有的计算都是在Gaussian 09程序包[13]下完成.
2 结果和讨论
2.1 几何构型
表1列出了配合物1和2在B3LYP/6-31+G(d)-LANL2DZ水平下优化得到的主要键长、键角和二面角参数,图1画出了配合物1和2的几何结构.由表1和图1可知,配合物1和2呈现四配位的平面构型,这与实验报道的结果[4]一致.在配体的对位引入-Br或-NO2对配合物1和2结构的影响很小,它们具有相似的键长Ni-N1、Ni-N2、Ni-O1和Ni-O2,且键角N1-Ni-O2和N2-Ni-O1及二面角N1-N2-O1-O2都接近于180°.
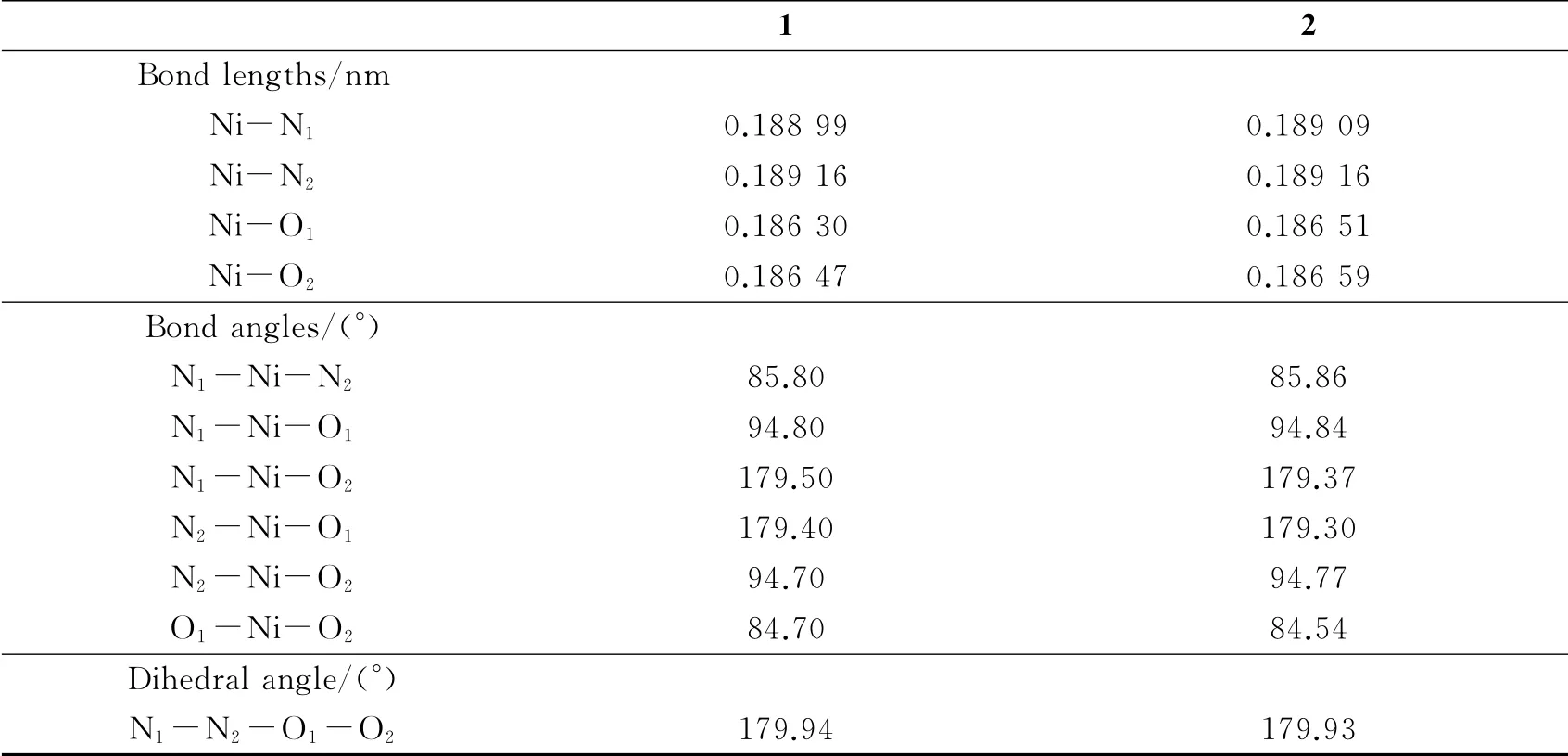
表1 在B3LYP/6-31+G(d)-LANL2DZ水平下优化的配合物1和2的几何参数Table 1 The optimized geometric parameters of complexes 1 and 2 at the B3LYP/6-31+G(d)-LANL2DZ level
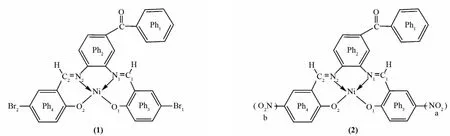
图1 配合物1和2的几何结构Fig.1 The geometric structures of complexes 1 and 2
2.2 前线分子轨道
前线分子轨道,包括最高占据轨道(HOMO,H)和最低未占据轨道(LUMO,L),对研究电子吸收光谱性质具有重要作用.表2列出了配合物1和2在基态的分子轨道分布.由表2知,配合物1和2占据轨道的分布特点大致相同.例如,两者H-1轨道主要分布在Ph2、Ph3和Ph4基团上,H-3轨道主要分布在金属Ni原子上.但是它们LUMOs轨道的电子云分布差别较大.配合物1的L+2轨道主要分布在金属Ni原子上,而配合物2的L+2轨道主要由Ph4和-NO2基团组成.图2绘出了这两种配合物的部分前线分子轨道能级,并给出了几个在吸收光谱中涉及到的重要分子轨道轮廓图.由图2知,席夫碱对位上取代基吸电子性的不同对HOMO轨道的影响大于对LUMO轨道的影响.当在席夫碱的对位引入更强的吸电子基团-NO2时,席夫碱配体成为一个强的电子受体,从而降低了配合物2的HOMO轨道能量.然而LUMO轨道的能量几乎不受影响,因此导致了H-L能级差减小.
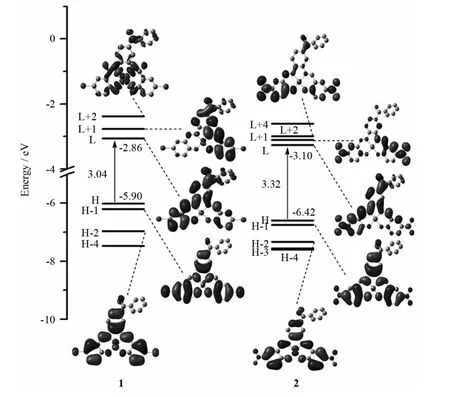
图2 吸收光谱中配合物1和2的前线分子轨道轮廓图、能级及能级差Fig.2 Presentation of the profiles,energy levels,energy gaps of frontier molecular orbitals involved in the absorption spectra for complexes 1 and 2
2.3 吸收光谱
在优化的几何结构的基础上,采用TD-B3LYP/6-31+G(d)-LANL2DZ方法,计算了配合物1和2在N,N-二甲基甲酰胺溶液中的紫外吸收光谱,拟合所得的吸收光谱如图3和表3所示.从图3可以看出,在320~500nm的区间内,配合物1和2都存在4个强度不同的吸收峰.由表3知,两个配合物的最长吸收波长分别位于619和615nm处.与配合物1相比,配合物2的最大吸收波长呈现出蓝移的现象,这与它们HL能级差的变化规律是一致的.配合物1的最强吸收峰位于363nm处(3.42eV),主要由H-2→L(91%)的跃迁组成.由于H-2轨道主要分布在Ni原子及Ph2,Ph3和Ph4基团上,而L轨道主要分布在Ph2,Ph4和CO基团及C2原子上,所以该跃迁归属为(d,π)→(p,π*),且该吸收峰主要为ILCT/MLCT性质.配合物2的最强吸收峰位于426nm处(2.91eV),该峰是由H-1→L(83%)跃迁组成的,具有ILCT性质.从表3和图3还可以看出,理论计算得到的峰值与实验结果吻合得较好,并且还得到了实验上没有测得的吸收峰.例如配合物1和2分别在325nm(振子强度f=0.321 3)和426nm(f=0.618 9)拟合到了强的理论峰,但实验上对此却没有进行报道.
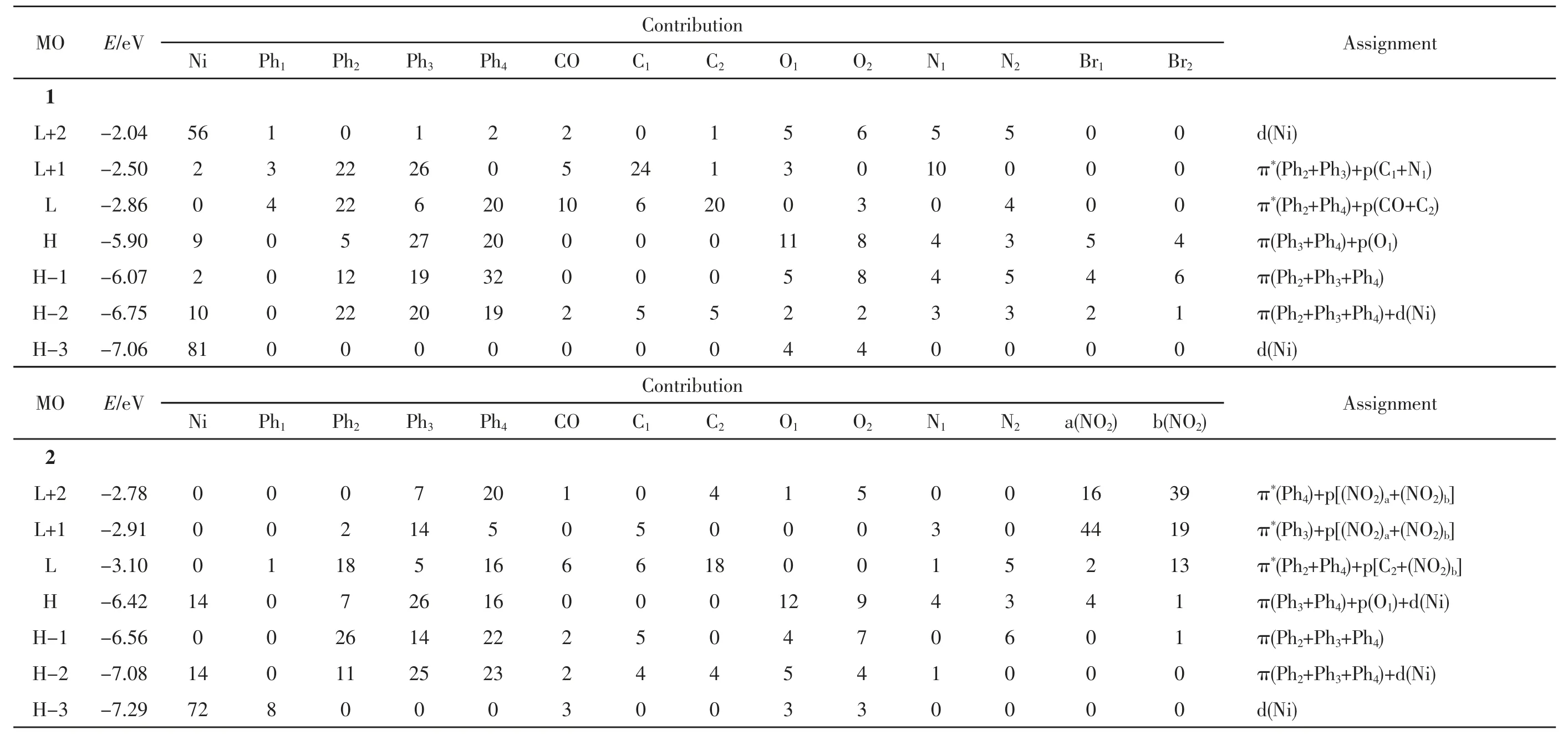
表2 在B3LYP/6-31+G(d)-LANL2DZ 水平下配合物1和2在DMF中基态分子轨道电子云分布Table 2 Molecular orbital compositions in the ground state of complexes 1 and 2 at B3LYP/6-31+G(d)-LANL2DZ level in DMF
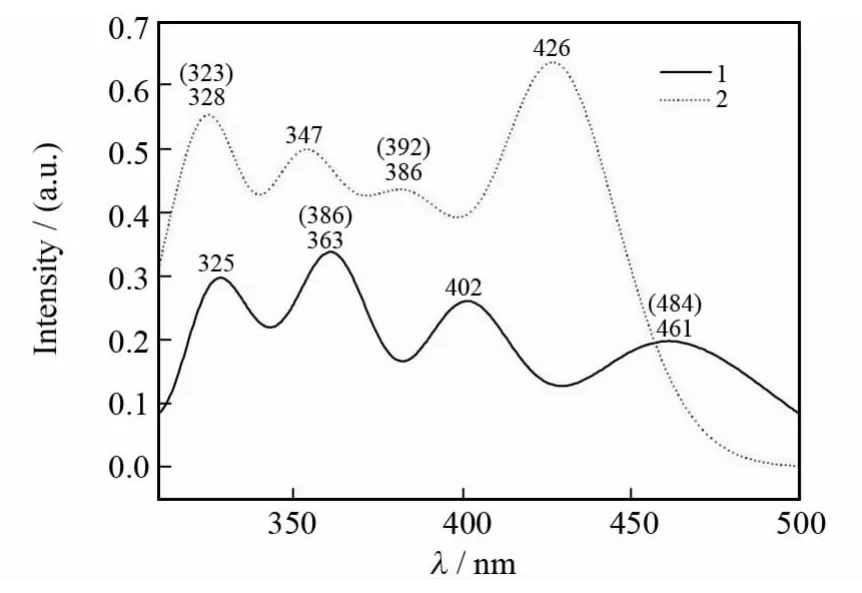
图3 在TD-B3LYP(PCM)水平下配合物1和2在N,N-二甲基甲酰胺溶液中的吸收光谱曲线及相应的实验数据(括号中)Fig.3 Simulated absorption spectra in N,N-dimethylformamide for complexes 1 and 2 from the TD-B3LYP(PCM)calculation together with experimental values in parentheses
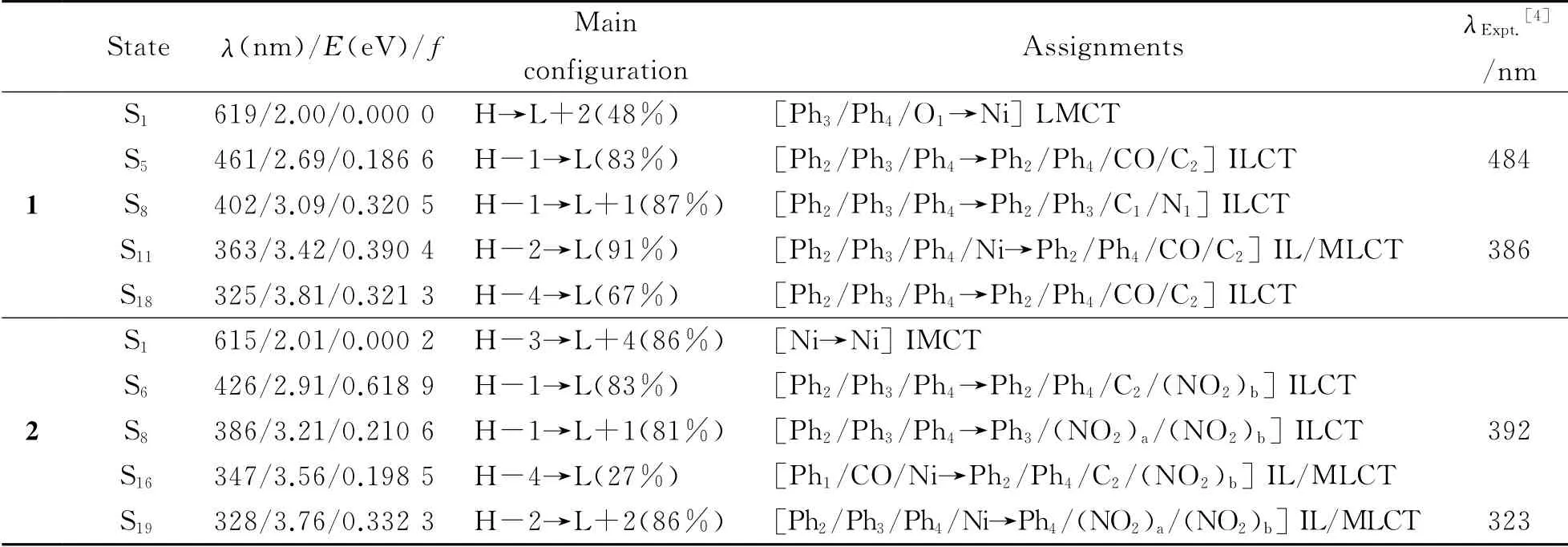
表3 在TD-B3LYP水平下配合物1和2在N,N-二甲基甲酰胺溶剂中的吸收光谱数据以及相应的实验值Table 3 Calculated absorptions of the complexes 1 and 2 in N,N-dimethylformamide medium at TD-B3LYP level together with experimental values
3 结论
在B3LYP/6-31+G(d)-LANL2DZ水平下优化了配合物1和2的几何构型,用含时密度泛函方法(TDDFT)计算了它们在N,N-二甲基甲酰胺溶液中的吸收光谱,并探讨了它们的电子跃迁性质.计算结果表明在席夫碱配体的对位引入-Br或-NO2对配合物1和2的几何结构影响很小.但是对位处的-NO2由于强吸电子性增大了配合物2 HOMO-LUMO的能级差,使其最大吸收波长相对于配合物1发生了蓝移.
[1]KOCYIGIT O,KURSUNLU A N,GULER E.Complexation properties and synthesis of a novel Schiff base with triphenylene nucleus[J].J Hazard Mater,2010,183(1):334-340.
[2]高 敏,杨天林,马志强,等.3,5二碘水杨醛缩水杨酰肼Schiff碱及其金属配合物的合成与抑菌活性[J].应用化学,2011,28(10):1167-1172.
[3]ISMAIL T M A,SALEH A A,GHAMRY M A E.Tetra-and hexadentate Schiff base ligands and their Ni(II),Cu(II)and Zn(II)complexes.Synthesis,spectral,magnetic and thermal studies[J].Spectrochim Acta Part A,2012,86:276-288.
[4]ASADI M,SEPEHRPOUR H,MOHAMMADI K.Tetradentate Schiff base ligands of 3,4-diaminobenzophenone:Synthesis,characterization and thermodynamics of complex formation with Ni(II),Cu(II)and Zn(II)metal ions[J].J Serb Chem Soc,2011,76(1):63-74.
[5]BECKE A D.Density-functional thermochemistry.III.The role of exact exchange[J].J Chem Phys,1993,98(7):5648-5652.
[6]LEE C,YANG W,PARR R G.Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988,37:785-789.
[7]LV P C,LI H Q,SUN J,et al.Synthesis and biological evaluation of pyrazole derivatives containing thiourea skeleton as anticancer agents[J].Med Chem,2010,18(13):4606-4614.
[8]ELSABEE M Z,SABAA M W,NAGUIB H F,et al.Polymerization and chelating behavior of N-acryloyl,N-phenylthiourea[J].Polym Bull,1989,22:143-149.
[9]WADT W R,HAY P J.Ab initio effective core potentials for molecular calculations.Potentials for main group elements Na to Bi[J].J Chem Phys,1985,82(1):284-298.
[10]HAY P J,WADT W R.Ab initio effective core potentials for molecular calculations.Potentials for K to Au including the outermost core orbitals[J].J Chem Phys,1985,82(1):299-310.
[11]MIERTUS S,SCROCCO E,TOMASI J.Electrostatic interaction of a solute with a continuum.A direct utilizaion of ab initiomolecular potentials for the prevision of solvent effects[J].Chem Phys,1981,55(1):117-129.
[12]TOMASI J,PERSICO M.Molecular interactions in solution:an overview of methods based on continuous distributions of the solvent[J].Chem Rev,1994,94(7):2027-2094.
[13]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09[CP].Revision A.02,Gaussian,Inc.,Wallingford CT,2009.
