Theoretical studies on the structure and vibrational spectra of some tri(4-nitrophenyl)methane derivatives
2014-11-27WUWenpengCAOYan
WU Wenpeng,CAO Yan
(1.College of Chemistry and Chemical Engineering,Institute of Environmental and Analytical Sciences,Henan University,Kaifeng 475004,Henan,China;2.Xinjiang Hetian No.2 Middle School,Hetian 848000,Xinjiang,China)
Triphenylmethane(TPM)derivatives are a kind of important complexes with a very wide range of application.For example,they can be used as dyes[1-4],fluorescent probes[5],atom transfer radical polymerization initiators[6-7],inhibitors of Hepatitis C virus helicase[8]and anticancer agent[9].It has also been reported that TPM,as a possible moderator material,might find promising applications in cold neutronmoderator[10].
Recently,XU et al synthesized a series of TPM derivatives and characterized their chemical structures based on infrared(IR)spectrometry[11-14].However,they only assigned a few peaks in the IR spectra while leaving most of the peaks unassigned.Therefore,in the present research,we adopt density functional theory(DFT)method to theoretically study and assign the vibrational spectra of triphenylmethane(1in Fig.1)and three 4-nitro group substituted TPM derivatives(2,3 and 4 in Fig.1)in order to gain more insights into the vibrational spectra.We focus on the theoretical calculations with DFT method,since this method is powerful in predicting the geometry[2,15-21]and vibrational frequency[2,15-19],which provides good agreement with experimental data.Thus the vibrational spectra(including IR spectra and Raman spectra)of 1,2,3 and 4 were calculated with DFT method,and the assignments of the spectra were made based on the calculations.
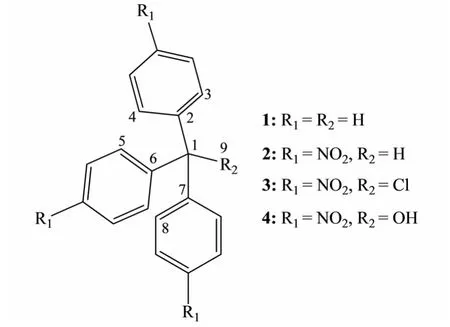
Fig.1 The skeleton structure of TPM derivatives
1 Computational details
The geometry was optimized with DFT-B3LYP[22-23]functional and 6-31G*basis set.The vibrational frequencies were calculated at the same level to obtain the IR and Raman spectra.Computing results show that all the frequencies are real,which indicate that all the structures optimized are stable ones.Then all the frequencies are scaled by a factor of 0.96 to compare with the experimental ones.All the above calculations were performed with Gaussian09 program[24].
2 Results and discussion
2.1 Geometry
Some of the calculated structural parameters are listed in Table 1.It can be seen that 1-2bond lengths(the numbers 1and 2here are the atomic numbers shown in Fig.1,the same below)are almost the same when para-H in benzene is substituted by-NO2(1→2),but the 1-2bond is slightly elongated when the H atom in the centered-C is substituted by Cl atom or-OH(2→3,4).This implies that the stretching vibration frequency of the C-C bond in 3 and 4 is larger than that in 1 and 2.As to 2,3 and 4,the bond lengths of C-N and N-O are almost the same,which means that they exhibit similar stretching vibration frequencies.Besides,the bond angles and dihedral angles of 1 and 2 are similar,while bond angles and dihedral angles of 3 are of a little deviation as compared with those of 1 and 2.As compared with CH4,the bond angle 2-1-6in 1 and 2 tends to increase,which is because the larger steric hindrance effect between the benzene rings results in decreasing in bond angle 2-1-9.Moreover,the bond angle 2-1-9 in 3 is a little bit larger than that in 2,which may be because Cl atom has larger electronegativity than H and there is larger repulsion between Cl and benzene ring,leading to smaller bond angel 2-1-6.In addition,due to theintroduction of-OH,the asymmetry of 4increases and three series of data appear because of the presence of three benzene rings in 4.The difference mainly lies in the dihedral angles(See details in Table 1).
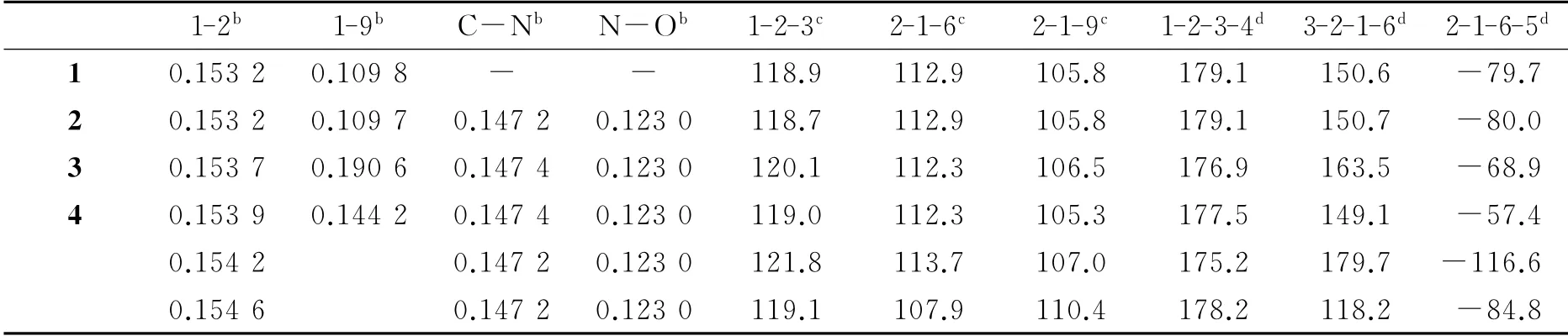
Table 1 Selected geometric parameters of compounds 1-4a
2.2 IR spectra
The simulated IR spectra of 1-4 are shown in Fig.2,and the assignments of the peaks are listed in Tables 2-5,while available experimental values and experimental assignments are also provided for comparisons.It can be seen that the calculated values show good accordance with the experimental ones.Namely,the frequencies of C-N(or-NO2)stretching vibration in 2,3 and 4 are almost the same,which are in accordance with the geometry discussed above.Particularly,it might be worth pointing out that the experiments provide only 7assigned peaks of 3 and 4[14],but herein we have assigned most of the peaks in the IR spectra.Furthermore,in combination with relevant theoretical calculations(abridged as Calc.),we suppose that some experimental(abridges as Expt.)assignments may be inappropriate.For example,the calculated assigned peak of ring butterfly vibration at 517cm-1in 3 is assigned as C-Cl stretching in the experiment;the calculated assigned peak at 841cm-1both in 3 and 4,referring to-NO2scissoring,ring out of plane bending and centered-C bending vibration,is assigned as C-N stretching in the experiment.In fact,C-Cl stretching vibration peaks appear around 340cm-1(Calc.,out of the range of Expt.)and 700-800cm-1(Expt.)in association with other vibration modes,while C-N stretching vibration peaks occur at 1 110cm-1and 1 350cm-1(Expt.)in association with other vibrations.
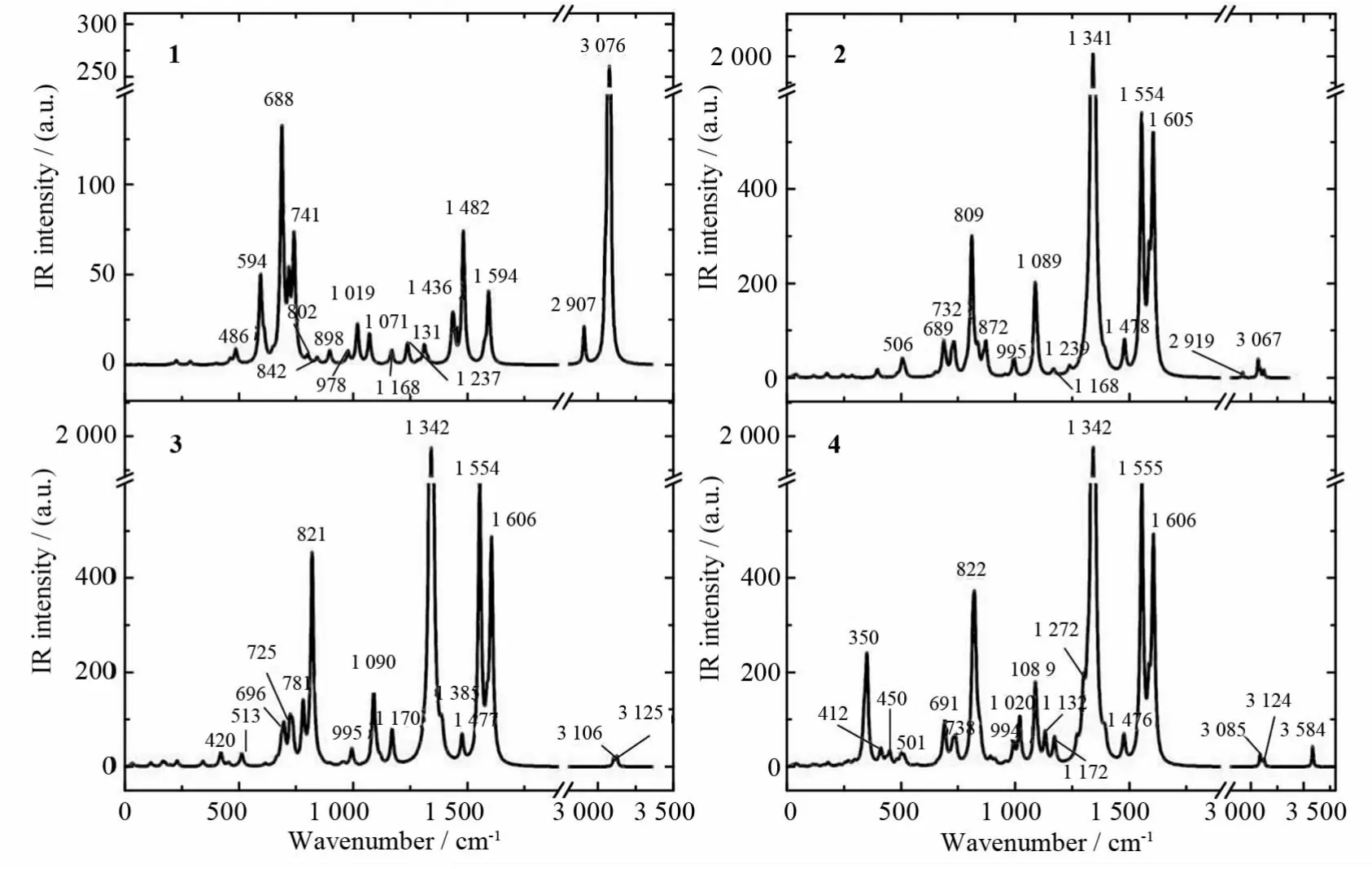
Fig.2 The theoretically simulated IR spectra of compounds 1-4
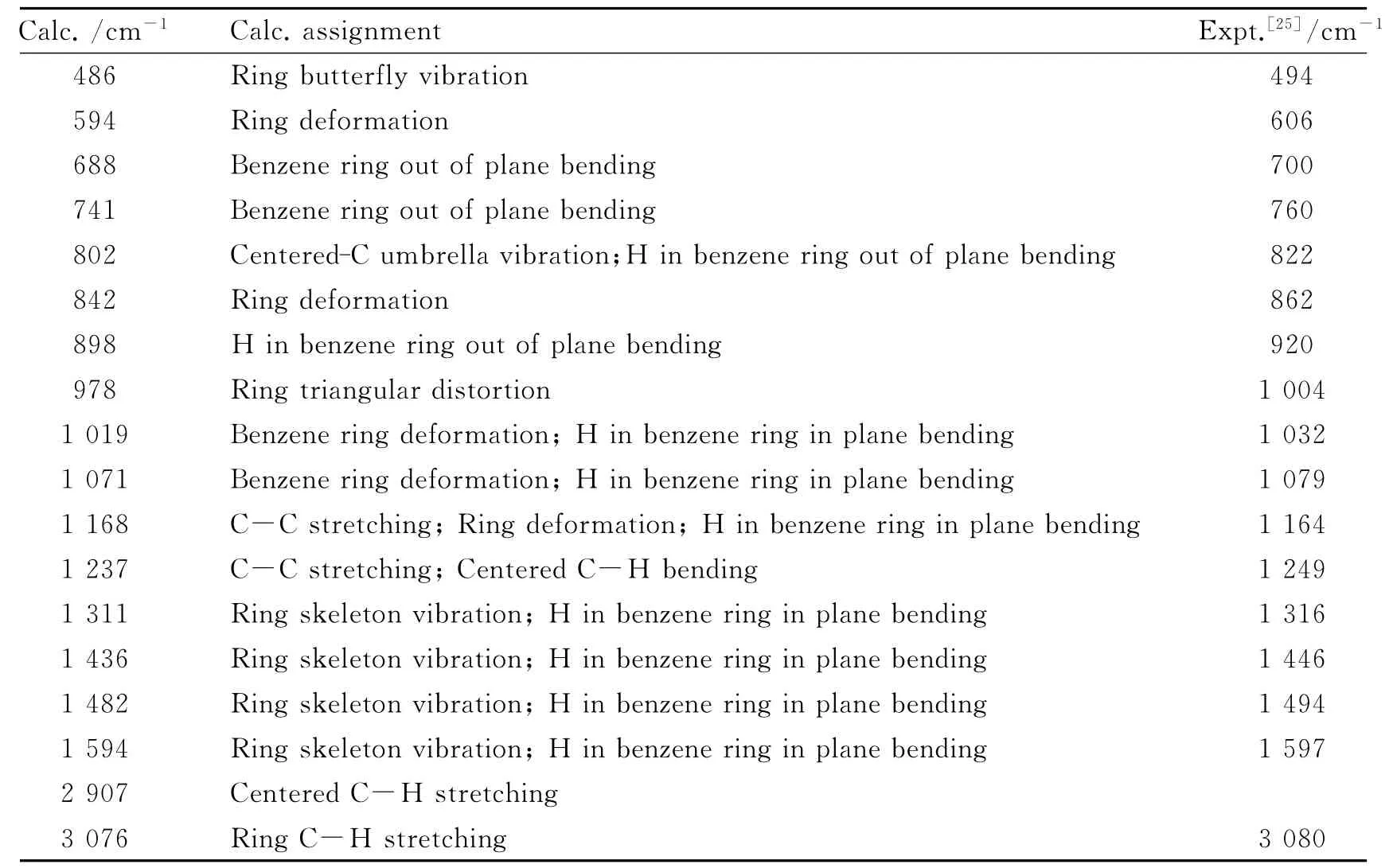
Table 2 The assignment of IR spectrum of 1

Table 3 The assignment of IR spectrum of 2
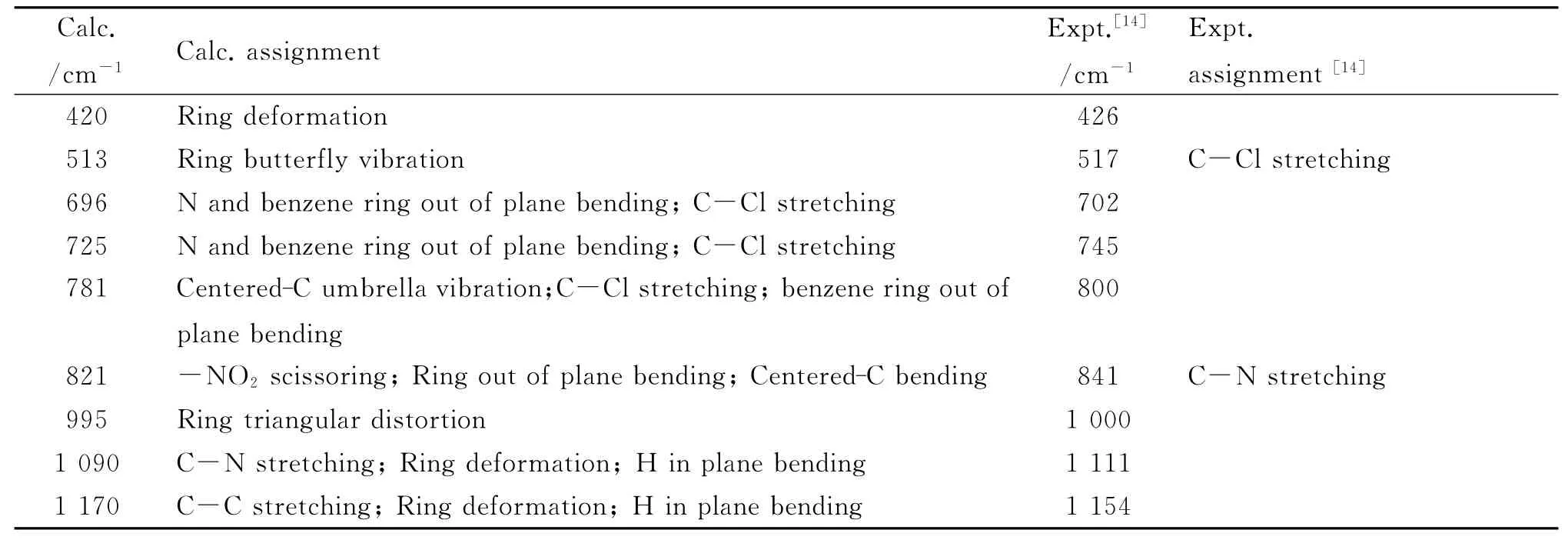
Table 4 The assignment of IR spectrum of 3

Continued to Table 4
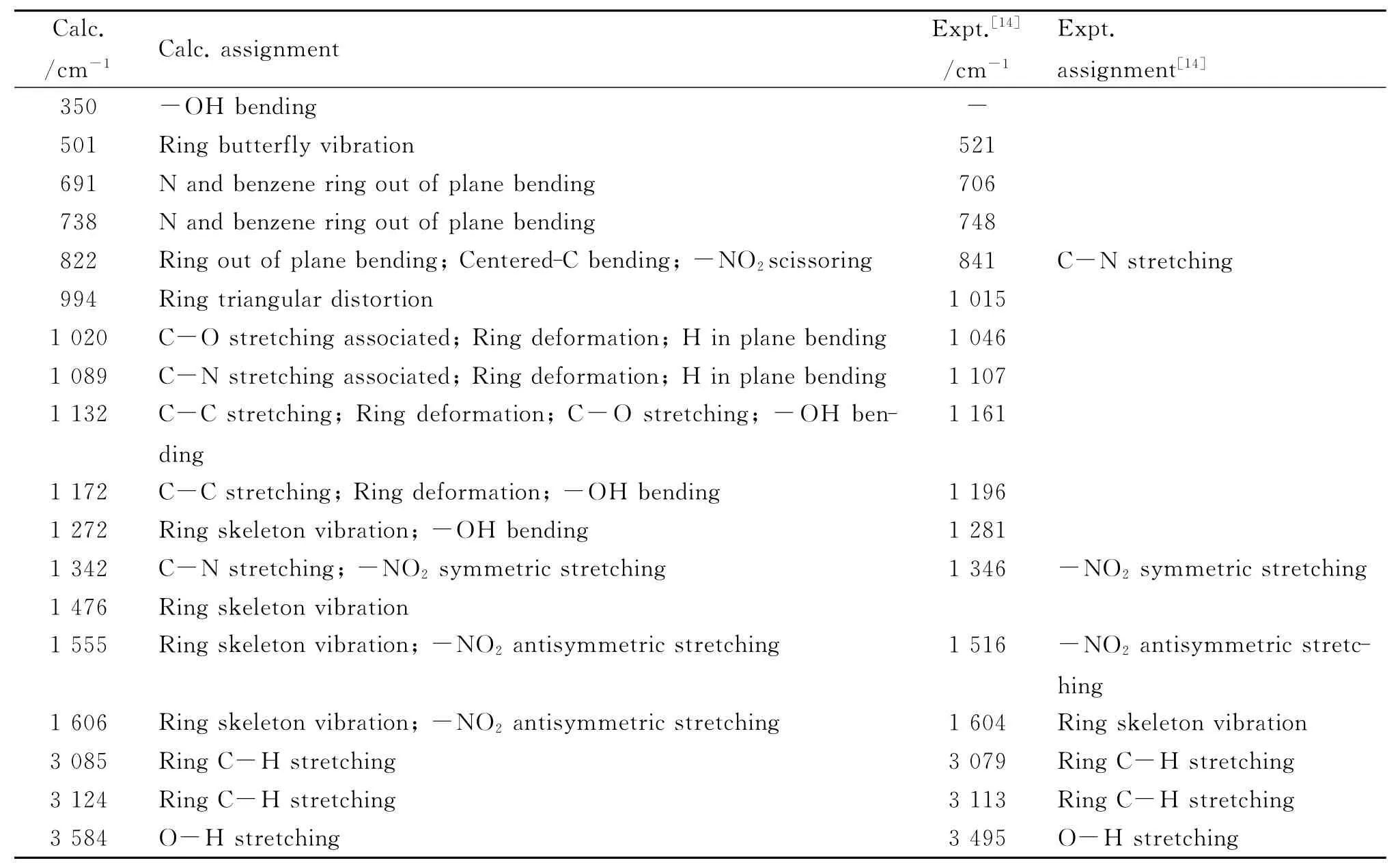
Table 5 The assignment of IR spectrum of 4
2.3 Raman spectra
The simulated Raman spectrum of 1is shown in Fig.3.The assignments of the Raman peaks are listed in Table 6in comparison with the experimental values[25].It can be seen that the calculated values agree well with the experimental ones,which indicates that the theoretical method used here is reliable.
In the meantime,we also calculated the Raman spectra of 2,3 and 4 with the same method,and the results are shown in Fig.3 and Table 7.Comparing 2 with 1,we can see that two additional peaks of 2 appear at 1 088cm-1and 1 344cm-1,and they can be assigned to C-N stretching coupled with C-H in plane bending vibration and C-N stretching coupled with-NO2symmetric stretching vibration,respectively;Moreover,the ring skeleton vibration shifts to red,while C-H(both centered and ring C-H)stretching modes shift to blue.From the Raman spectra of 2,3 and 4,it can be seen that they are similar to each other and the main difference is that centered C-H stretching(2 919cm-1)appears only in 2,while O-H stretching(3 584cm-1)appears only in 4,which can be used to distinguish these three compounds.
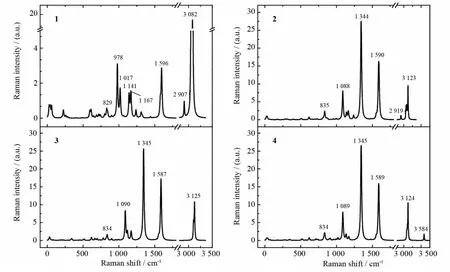
Fig.3 The theoretically simulated Raman spectra of compounds 1-4

Table 6 The assignment of Raman spectrum of 1

Table 7 The assignment of Raman spectra of 2-4
3 Conclusions
The geometries of triphenylmethane 1 and its derivatives 2,3 and 4 were optimized by DFT method,and their IR and Raman spectra were calculated therewith.We not only made assignments of the peaks in the experimental spectra,but also corrected some inappropriate assignments in Ref.[14].For instance,the IR peak at 517cm-1in 3 should be assigned to ring butterfly vibration rather than C-Cl stretching;that at 841cm-1in3 and 4 should be assigned to-NO2scissoring,ring out of plane bending and centered-C bending vibration rather than C-N stretching vibration.In the meantime,although the simulated Raman spectra of 2,3 and 4 are similar to each other,they can still be distinguished by the peaks at 2 919 cm-1and 3 584cm-1.Namely,the peak at 2 919cm-1is assigned to the centered C-H stretching vibration only in 2,while that at 3 584cm-1is assigned to the O-H stretching vibration only in 4.
[1]DUXBURY D F.The photochemistry and photophysics of triphenylmethane dyes in solid and liquid media[J].Chem Rev,1993,93:381-433.
[2]BARDAJEE G R.SbCl3-catalyzed one-pot synthesis of 4,4′-diaminotriarylmethanes under solvent-free conditions:Synthesis,characterization,and DFT studies[J].Beilstein J Org Chem,2011,7:135-144.
[3]HAMMERSHØJ P,SØRENSEN T J,HAN B H,et al.Base-assisted one-pot synthesis of N,N′,N″-triaryltriazatriangulenium dyes:enhanced fluorescence efficiency by steric constraints[J].J Org Chem,2012,77:5606-5612.
[4]THYRHAUG E,SØRENSEN T J,GRYCZYNSKI I,et al.Polarization and symmetry of electronic transitions in long fluorescence lifetime triangulenium dyes[J].J Phys Chem A,2013,117:2160-2168.
[5]GUO J H,ZHU L N,KONG D M,et al.Triphenylmethane dyes as fluorescent probes for G-quadruplex recognition[J].Talanta,2009,80(2):607-613.
[6]XU Y Q,LU J M,XU Q F,et al.Atom transfer radical polymerization of styrene initiated by triphenylmethyl chloride[J].Europ Polym J,2005,41:2422-2427.
[7]XU Y Q,XU Q F,LU J M,et al.Reverse atom transfer radical polymerization of MMA initiated by triphenylmethane[J].Polymer Bulletin,2007,58:809-817.
[8]CHEN C S,CHIOU C T,GRACE S C,et al.Structure-based discovery of triphenylmethane derivatives as inhibitors of Hepatitis C virus helicase[J].J Med Chem,2009,52(9):2716-2723.
[9]PALCHAUDHURI R,NESTERENKO V,HERGENROTHER P J.The complex role of the triphenylmethyl motif in anticancer compounds[J].J Am Chem Soc,2008,130:10274-10281.
[10]HÜGLE T,MOCKO M,HARTL M A,et al.Triphenylmethane,apossible moderator material[J].Nucl Instrum Methods Phys Res A,2014,738:1-5.
[11]徐元清,丁 涛,房晓敏,等.三(对乙酰基苯基)甲烷的合成及表征[J].化学研究,2008,19(2):19-21.
[12]徐元清,丁 涛,房晓敏.三苯甲烷-4,4′,4″-三羧酸的合成[J].化学研究,2011,22(1):26-28.
[13]李武斌,徐元清,张延兵,等.三苯甲醇-4,4′,4″-三羧酸及氯化物的合成[J].化学研究,2012,23(5):8-11.
[14]蒋清民,徐元清,卞龙骧,等.三(4-硝基苯基)甲醇及其氯化物的合成与表征[J].化学研究,2014,25(4):378-380.
[15]JOSEPH L,SAJAN D,CHAITANYA K,et al.Molecular conformational analysis,vibrational spectra and normal coordinate analysis of trans-1,2-bis(3,5-dimethoxy phenyl)-ethene based on density functional theory calculations[J].Spectrochim Acta A,2014,122:375-386.
[16]LI R,JI W,CHEN L,et al.Vibrational spectroscopy and density functional theory study of 4-mercaptophenol[J].Spectrochim Acta A,2014,122:698-703.
[17]SYLVESTRE S,SEBASTIAN S,EDWIN S,et al.Vibrational spectra(FT-IR and FT-Raman),molecular structure,natural bond orbital,and TD-DFT analysis of L-asparagine monohydrate by density functional theory approach[J].Spectrochim Acta A,2014,133:190-200.
[18]郑燕升,卓志昊,李军生.白花丹素分子结构和红外光谱的密度泛函理论研究[J].化学研究,2011,22(3):61-65.
[19]KERN B,STRELNIKOV D,WEIS P,et al.IR,NIR,and UV absorption spectroscopy of C602+and C603+in neon matrixes[J].J Phys Chem Lett,2014,5:457-460.
[20]刘 霞,王献伟,赵高峰,等.GemSin(m=1,2;n=1~7)团簇结构与性质的密度泛函理论研究[J].河南大学学报:自然科学版,2008,38(1):17-21.
[21]宁 攀,赵建想.利用密度泛函理论研究α-联噻吩体系H(C4H2S)nH的结构和电子光谱[J].化学研究,2013,24(5):493-500.
[22]BECKE A D.Density-functional thermochemistry.III.The role of exact exchange[J].J Chem Phys,1993,98(7):5648-5652.
[23]LEE C,YANG W,PARR R G.Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988,37:785-789.
[24]FRISH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09[CP].Revision A.02,Gaussian,Inc.,Wallingford CT,2009.
[25]National Institute of Advanced Industrial Science and Technology.Spectral database for organic compounds SDBS[DB/OL].2014-05-30.http://sdbs.db.aist.go.jp.
