鸟氨酸氨基甲酰转移酶缺陷病1例病例报告
2014-11-22朱燕凤王慧君吴冰冰谢新宝
朱燕凤 王慧君 吴冰冰 谢新宝 陆 怡 俞 蕙
1 病例资料
女,5岁。因“反复呕吐2月余伴肝功能异常1月半”于2014年3月3日入复旦大学附属儿科医院。病程中患儿无发热、寒战和抽搐等表现。
患儿既往体健,无肝病史及类似病史,无血制品输注史。其父母及一姐姐均体健,无类似病史,父母否认近亲结婚,否认家族遗传疾病史。
图3为患儿起病后的症状、体征、诊断和治疗等重要临床信息时间轴。
2 讨论
先天性高氨血症是一种较少见的先天性代谢异常疾病,是由于尿素合成的代谢异常,导致血氨增高,最终造成神经系统功能损害的疾病。其最常见的病因是尿素循环障碍。尿素循环过程包括5个步骤,共有6个酶参与,其中任何一个酶缺乏均可使尿素循环出现减少或终止,故临床上将其分为6种类型:氨基甲酰磷酸合成酶(CPS)Ⅰ缺陷、N-乙酰谷氨酸合成酶缺陷、OTC缺陷、精氨酸代琥珀酸合成酶缺陷、精氨酸代琥珀酸裂解酶缺陷和精氨酸酶缺陷[1]。
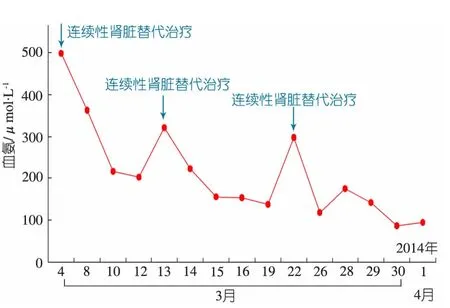
图2 本文病例血氨变化
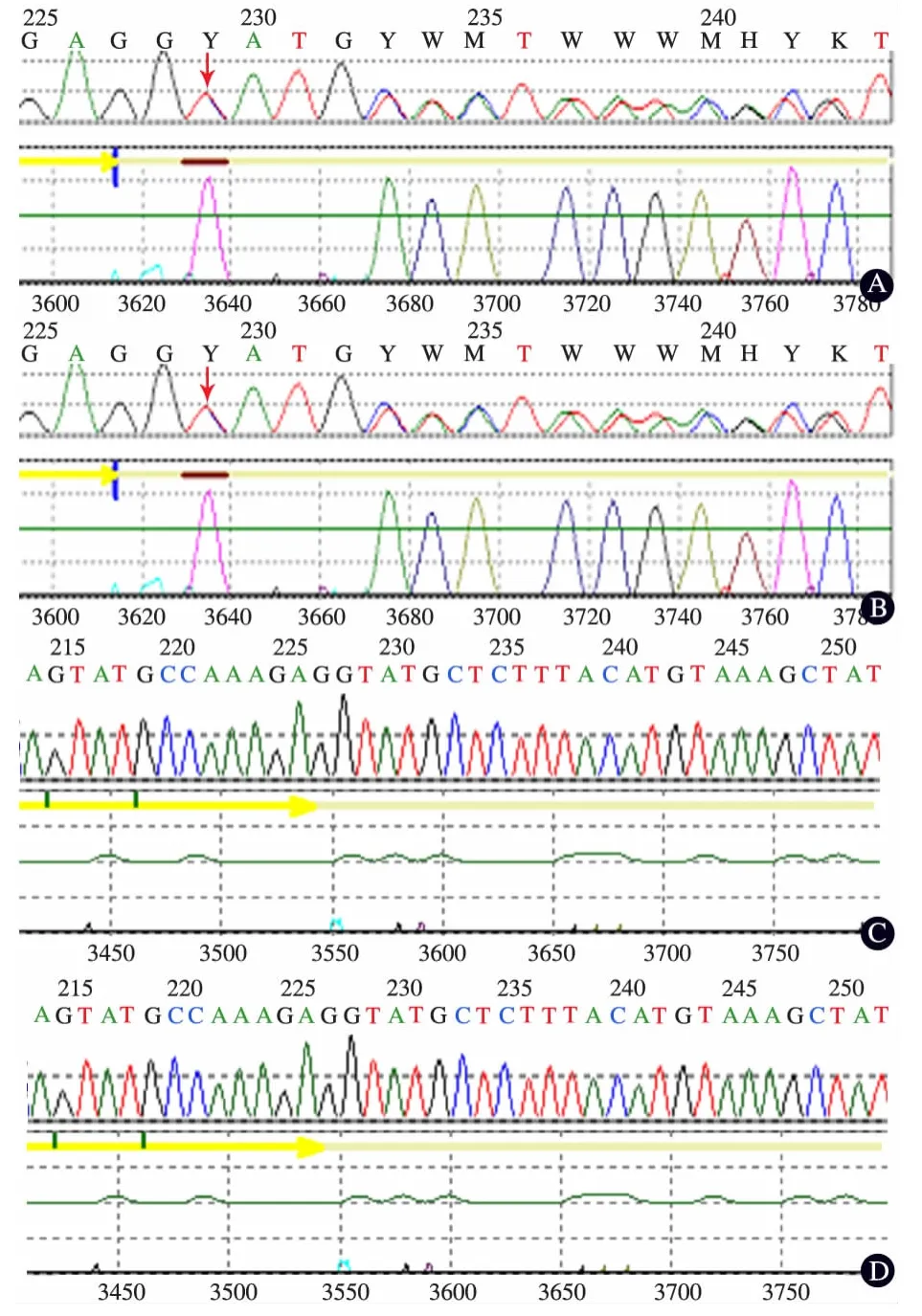
图3 本文病例及其家庭成员OTC基因测序结果
OTC是尿素循环关键酶之一,催化鸟氨酸和氨基甲酰磷酸生成瓜氨酸。OTC缺陷病(OTCD)是先天性尿素循环障碍中常见的类型,多为不完全性显性或隐性X连锁遗传病[2]。在人群中的发病率为 1∶14 000[3]。根据起病时间将OTCD分为2种类型:新生儿急性起病型和新生儿后起病型(迟发型)。本文患儿生长发育正常,5岁时以呕吐为首发症状就诊,属于迟发型。食欲不佳及呕吐是高氨血症最显著的首发表现,年龄大者可出现共济失调、癫 、情绪不稳定、攻击性行为和智力低下等,甚至出现昏迷[1]。本文患儿的临床表现较典型,随着血氨的升高(最高达498μmol·L-1),临床症状加重。高氨血症患儿临床表现与血氨浓度密切相关,血氨>100μmol·L-1可表现为兴奋及行为异常,200μmol·L-1左右可出现意识障碍和惊厥,>400μmol·L-1可出现昏迷、呼吸困难,甚至猝死[4,5]。
OTCD为X连锁显性遗传,男性病情常较女性重,除了血氨增高及肝功能异常外,血尿串联质谱有特征性的改变,主要表现为高鸟氨酸血症、低瓜氨酸血症、尿乳清酸增高[4,5]。本文患儿2次尿串联质谱中,仅1次尿乳清酸增高,为患儿经连续性肾脏替代治疗后血氨下降时,在药物和饮食控制治疗3 d后测血尿串联质谱检查正常。血尿氨基酸的检查对诊断OTCD非常重要,但阴性不能排除本病,在急性发作期送检标本可提高阳性率。根据血尿氨基酸及尿有机酸的水平,还可鉴别引起高氨血症尿素循环4种酶的缺陷[1,6,7]:血瓜氨酸降低、尿乳清酸升高为 OTCD;血瓜氨酸升高、尿乳清酸升高为瓜氨酸血症Ⅰ型(精氨酰琥珀酸合成酶缺乏症);血瓜氨酸升高、血精氨酸及尿乳清酸正常为瓜氨酸血症Ⅱ型;血精氨酸升高为精氨酸血症(精氨酸酶缺乏症)。
氨对神经系统与肝脏均有很强的毒性作用,当患儿血氨水平>200μmol·L-1,且排除其他疾病的情况下可以考虑高氨血症。大脑中的氨可被合成为谷氨酰胺以达到解毒的目的。当血氨浓度明显增高时,谷氨酰胺大量合成,导致大脑中的α-酮戊二酸被大量消耗。作为三羧酸循环的重要中间产物,α-酮戊二酸缺乏会导致三羧酸循环障碍,从而使神经系统能量代谢出现障碍,出现脑水肿,严重时会引发颅内高压、肝昏迷,并最终导致死亡[1]。本文患儿虽然入院前头颅MRI及入院后头颅CT无明显异常,但EEG示脑电活动明显变慢,提示神经系统受累。除了脑部病变,肝脏亦是主要的受累器官,OTC杂合子的女性则有肝肿大和不同的病理学变化,如脂肪变、肝炎症病变、肝纤维化及糖原沉积等,电镜下可见内质网的扩张,线粒体的弯曲及分支[8]。本文患儿肝穿刺病理示肝细胞内糖原凝聚块形成,门管区纤维轻度增生伴个别纤维间隔形成,符合OTC杂合子的女性肝脏病理改变。
OTCD目前可以进行基因水平的诊断,该基因位于Xp21.1,已在人类OTC基因中发现了341处突变与疾病相关[9],OTC基因10个外显子和9个内含子都存在突变,而外显子1和2突变比例较高。42%致病性突变与急性新生儿高氨血症有关,21%存在于迟发型病例,37%存在于女性杂合子。绝大多数新生儿型OTCD的突变涉及位于蛋白内部或活性位点的残基,而迟发型患者的突变大多与远离活性位点或位于蛋白表面的残基有关,多为外显子突变[10,11]。本文患儿及其母亲均测到OTC基因内含子7的杂合缺失突变(c.717+2T>T/C),该突变位点为已报道OTCD的致病突变位点,引起剪切突变,产生截短蛋白。同时发现患儿及其母亲OTC基因内含子7的杂合缺失插入突变(c.717+6_7het_indelTAATAATAG),未见文献报道,但HGMD(人类基因突变数据库)报道高氨血症的c.717+8_717+23del16突变[12],与本例发现的突变位置接近。患儿父亲样本未检测到该内含子区的突变。OTC基因位于X染色体,鉴于本文为女性患儿,母亲具有与之相同的杂合型突变,推测为父源性X染色体基因失活,导致该基因功能丧失。该夫妇如再生育,女孩有50%为正常,50%为携带者,由于X染色体失活是随机的,因此携带者女孩的临床表型将难以预测;男孩50%正常,50%为OCTD。可以为患儿母亲再孕时提供产前诊断参考。
OTCD的治疗以长期饮食疗法为主,低蛋白饮食是治疗的基础。可用药物建立代谢旁路以排出过多的氨,如精氨酸、苯甲酸钠和苯丁酸钠等。治疗期间应避免应激反应和感染,急性高氨血症昏迷者需行血液透析或腹膜透析和静注降血氨药物,以尽快清除过多的血氨。Leonard等[13]认为当血氨>500μmol·L-1就应进行积极干预,行血液滤过或腹膜透析治疗;新霉素和(或)乳果糖可减少肠道细菌产氨;肝移植是目前治疗OTCD的有效治疗方法,术后3年的生存率超过90%[14,15]。但肝移植手术时机的选择目前尚无统一的观点。由于高氨血症导致的神经系统损害是不可逆的,即使行肝移植术,术后部分受损的神经系统功能仍不能恢复正常[15,16]。此外,长期的饮食控制会导致患儿生长缓慢,也不利于肝移植的预后[17]。有学者主张在能较好地控制血氨的前提下,待患儿体重增长到8 kg时再行肝移植治疗,若患儿出现急性肝衰竭或药物治疗降血氨效果不佳,应立即行肝移植治疗[15]。
[1] Bachmann C.Mechanisms of hyperammonemia.Clin Chem Lab Med,2002,40(7):653-662
[2]Fantur M,Karall D,Scholl-Buergi S,et al.Recurrent somnolence in a 17-month-old infant:late-onset ornithine transcarbamylase(OTC)deficiency due to the novel hemizygous mutation c.535C>T(p.Leu179Phe).Eur J Paediatr Neurol, 2013, 17(1):112-115
[3]Wong DA.Ornithine transcarbamylase deficiency:are carrier females suitable donors?Pediatr Transplant, 2012, 16(6):525-527
[4]Nyhan WL, Rice-Kelts M, Klein J, et al.Treatment of the acute crisis in maple syrup urine disease.Arch Pediatr Adolesc Med,1998,152(6):593-598
[5]Matsuda I, Nagata N, Matsuura T, et al.Retrospective survey of urea cycle disorders:Part 1.Clinical and laboratory observations of thirty-two Japanese male patients with ornithine transcarbamylase deficiency.Am J Med Genet, 1991, 38(1):85-89
[6]Fukao T, Scriver CR, Kondo N.The clinical phenotype and outcome of mitochondrial acetoacetyl-CoA thiolase deficiency(beta-ketothiolase or T2 deficiency)in 26 enzymatically proved and mutation-defined patients.Mol Genet Metab,2001,72(2):109-114
[7]Yang YL(杨艳玲), Sun F,Qian N,et al.Clinical and laboratory screening studies on urea cycle defects.Chin J Pediatr(中华儿科杂志),2005,43(5):331-334
[8]周晓军,张丽华,主编.肝脏诊断病理学.南京:江苏科学技术出版社,2006.111-112
[9]Lopes-Marques M, Pereira-Castro I, Amorim A, et al.Characterization of the human ornithine transcarbamylase 3'untranslated regulatory region.DNA Cell Biol, 2012, 31(4):427-433
[10]Lin HY, Lin SP.Novel human pathological mutations.Gene symbol:OTC.Disease:ornithine transcarbamylase deficiency.Hum Genet, 2010, 127(4):475
[11]Brajon D, Carassou P, Pruna L, et al.Ornithine transcarbamylase deficiency in adult.Rev Med Interne, 2010,31(10):709-711
[12]Calvas P, Segues B, Rozet JM, et al.Novel intragenic deletions and point mutations of the ornithine transcarbamylase gene in congenital hyperammonemia.Hum Mutat,1998(S1):81-84
[13]Leonard JV, Morris AA.Urea cycle disorders.Semin Neonatol, 2002, 7(1):27-35
[14]Leonard JV, McKiernan PJ.The role of liver transplantation in urea cycle disorders.Mol Genet Metab,2004,81(S1):74-78
[15]Wakiya T, Sanada Y, Mizuta K, et al.Living donor liver transplantation for ornithine transcarbamylase deficiency.Pediatr Transplant, 2011, 15(4):390-395
[16]Wraith JE.Ornithine carbamoyltransferase deficiency.Arch Dis Child,2001,84(1):84-88
[17]McDiarmid SV,Merion RM,Dykstra DM,et al.Selection of pediatric candidates under the PELD system.Liver Transpl,2004,10(10 S2):23-30
