PTSD状态下HCN2-GLT-1在脊髓水平协同调控内脏高敏感性-中枢敏化的作用及机制研究
2014-11-05纪雷陈强何雨芩林玲冉亚梅朗秀琼李磊王建明王正国杨敏
纪雷,陈强,何雨芩,林玲,冉亚梅,朗秀琼,李磊,王建明,王正国,杨敏
创伤后应激障碍(post-traumatic stress disorder,PTSD)是指突发性、威胁性或灾难性事件导致个体延迟出现和长期持续存在的精神心理障碍[1-2]。既往研究证实,在PTSD状态下存在着广泛的内脏敏感性的改变,即痛觉敏化/去敏化现象[3-4],但其确切的分子机制尚不清楚。超极化激活环核苷酸门控阳离子通道(hyperpolarization-activated cyclic nucleotide-gated,HCN)的研究起源于超极化激活电流(hyperpolarization-activated current,Ih)的发现,有4种亚型,分别为HCN1、HCN2、HCN3和HCN4[5]。其中HCN2广泛分布于大脑、脊髓等神经组织,对神经元兴奋性高低及疼痛的调节起重要作用[6]。既往对HCN2在疼痛领域的研究主要集中在躯体痛方面,但有关HCN2在慢性内脏痛及应激相关的内脏高敏感性-中枢敏化中的作用及机制研究鲜见文献报道。
既往研究表明,谷氨酸在参与脊髓水平伤害性信息的传递及痛觉过敏中发挥着重要作用,是中枢神经系统最主要的兴奋性神经递质[7]。谷氨酸转运体-1(glutamate transporter-1,GLT-1)的激动剂头孢曲松(ceftriaxone,CTX)可选择性诱发编码GLT-1的基因转录,GLT-1可通过调节谷氨酸在突触前膜和突触后膜受体的利用率,双向调节突触内的谷氨酸水平及其功能活性,并可能通过影响HCN2离子通道的表达发挥抗伤害性刺激效应[8]。另外,HCN2的阻断剂(ZD7288)还可通过抑制突触前的Ca2+内流,调节突触前递质的释放[9-11]。为此,我们推测,在PTSD状态下,HCN2-GLT-1在脊髓水平协同调控内脏高敏感性-中枢敏化的形成中发挥了重要作用。本研究利用连续单一应激(SPS)联合足底电击刺激建立PTSD内脏高敏感大鼠模型,旨在阐明PTSD状态下HCN2-GLT-1在脊髓水平协同调控内脏高敏感性-中枢敏化的作用及机制。
1 材料与方法
1.1 PTSD内脏高敏感大鼠模型的建立及分组 选用清洁级成年雌性SD大鼠44只,体重150~200g,由第三军医大学大坪医院实验动物中心提供并经实验动物伦理委员会审查通过。随机分为3组:正常对照组(n=14)、PTSD内脏高敏感模型组(PTSD组,n=15)、PTSD+CTX组(n=15),每组中各有7只用于HCN2免疫荧光检测,其余用于内脏敏感性测定。正常对照组采用无干扰正常饲养;PTSD组按照文献[12]的方法,采用SPS联合足底电击刺激建立内脏高敏感大鼠模型,实验开始时将大鼠束缚2h,强迫游泳15min,休息15min,乙醚麻醉至意识丧失,清醒后给予1mA电击,持续4s,应激后7d开始实验。PTSD+CTX组除在PTSD术后给予CTX预处理(200mg/kg,腹腔注射)外,其余与PTSD组相同。
1.2 各组HCN2表达的免疫荧光检测 给药7d后的大鼠在深麻醉下用血管钳提起胸骨柄,剪开胸腔,暴露心脏,使用钝性针头插入左心室至升主动脉根部后,用血管钳固定;用0.01mol/L PBS溶液灌注,此时右心耳膨胀,用眼科剪小心剪开,让PBS溶液将整个大鼠的全身血液冲洗干净,直至肝脏颜色变白或从右心耳流出的液体变清为止,约需PBS溶液250ml。冲洗干净后,立即灌入4%多聚甲醛溶液(pH 7.4)约250ml固定30min以上;灌注完毕立即用血管钳咬开脊柱,取出脊髓腰段(L4-L6),4%多聚甲醛浸泡固定过夜,30%蔗糖溶液脱水过夜,0.01mol/L PBS溶液清洗后连续冠状切片,片厚25μm,以漂片法进行HCN2的免疫荧光染色。具体步骤如下:0.01mol/L PBS充分漂洗3次;加入封闭用正常羊血清工作液(北京中杉金桥生物技术有限公司),阻断非特异性免疫反应30min;加入一抗(兔抗HCN2,1:1000,Abcam公司),于4℃孵育过夜;0.01mol/L PBS漂洗3次;加入荧光二抗(FITC,1:200,北京中杉金桥生物技术有限公司)37℃孵育2h;0.01mol/L PBS漂洗3次;抗荧光淬灭封片剂(北京中杉金桥生物技术有限公司)封片。采用BX50 Olympus显微镜及Leica TCS SP2激光共聚焦显微镜观察并摄像,并采用Image-Pro Plus 6.0图像分析系统测量各组脊髓HCN2的平均光密度(A)值,表示HCN2的免疫荧光表达强度。
1.3 内脏敏感性的测定 参考文献[13]的方法,将大鼠按50mg/kg给予3%戊巴比妥腹腔注射麻醉,切开皮肤,将消毒电极埋植在一侧腹股沟韧带上方2cm处的腹外斜肌上,电极间隔约0.5~1.0cm,以丝线固定,电极另一端经皮下隧道引至颈背部并用橡胶套管保护后固定于皮肤,恢复后开始用于实验。采用生物机能信号采集系统记录结直肠扩张(colorectal distention,CRD)-内脏运动反射(visceromotor reflex,VMR),即通过CRD引起的腹外斜肌肌电活动(electromyogram,EMG)来评估内脏敏感性改变。参照既往文献[13],大鼠内脏敏感性测定前24h,禁食不禁水,在乙醚吸入麻醉下,将已埋植的电极与生物机能信号采集系统导线相连,由肛门插入扩张气囊,导管直径为2mm,气囊末端距肛门口约1cm,导管固定在鼠尾根部,待大鼠清醒并适应环境30min后行梯度结直肠扩张刺激(分别为10、20、40、60mmHg)。先以CRD压力为0mmHg时记录EMG 10s作为基线期,然后向球囊内快速充气,每一压力重复注气3次,维持20s,不同梯度压力间隔5min。诱发的EMG通过置于腹外斜肌的电极引出后,经放大、过滤和转换后录入计算机,使用SPIKE2分析软件计算平均曲线下面积(area under the curve,AUC)以定量EMG,并计算扩张期与基线AUC的差值,该AUC差值代表不同扩张压力的CRD诱导的VMR反应的程度。
1.4 统计学处理 采用SPSS 20.0软件进行统计学分析,计量资料用s表示,多组间比较采用单因素方差分析(ANOVA),进一步两两比较采用LSD-t法,P<0.05为差异有统计学意义。
2 结 果
2.1 PTSD状态下HCN2表达的变化及CTX对HCN2表达的影响 免疫荧光检测结果表明,与正常对照组比较,PTSD组脊髓HCN2表达明显增高(78.05±6.49 vs 121.12±4.85,P<0.001);与PTSD组比较,PTSD+CTX组HCN2表达显著降低(121.12±4.85 vs 98.24±5.86,P=0.012);与正常对照组比较,PTSD+CTX组HCN2表达亦显著升高(78.05±6.49 vs 98.24±5.86,P=0.024,图1)。
2.2 PTSD状态下内脏敏感性的改变 PTSD组AUCVMR在CRD为10、20mmHg时,与正常对照组比较差异无统计学意义,在CRD为40、60mmHg时明显高于正常对照组(P<0.001)。PTSD+CTX组AUCVMR在CRD为20、40、60mmHg时,均明显低于PTSD组(P<0.01),在CRD为40、60mmHg时,则明显高于正常对照组(P<0.01,表1)。
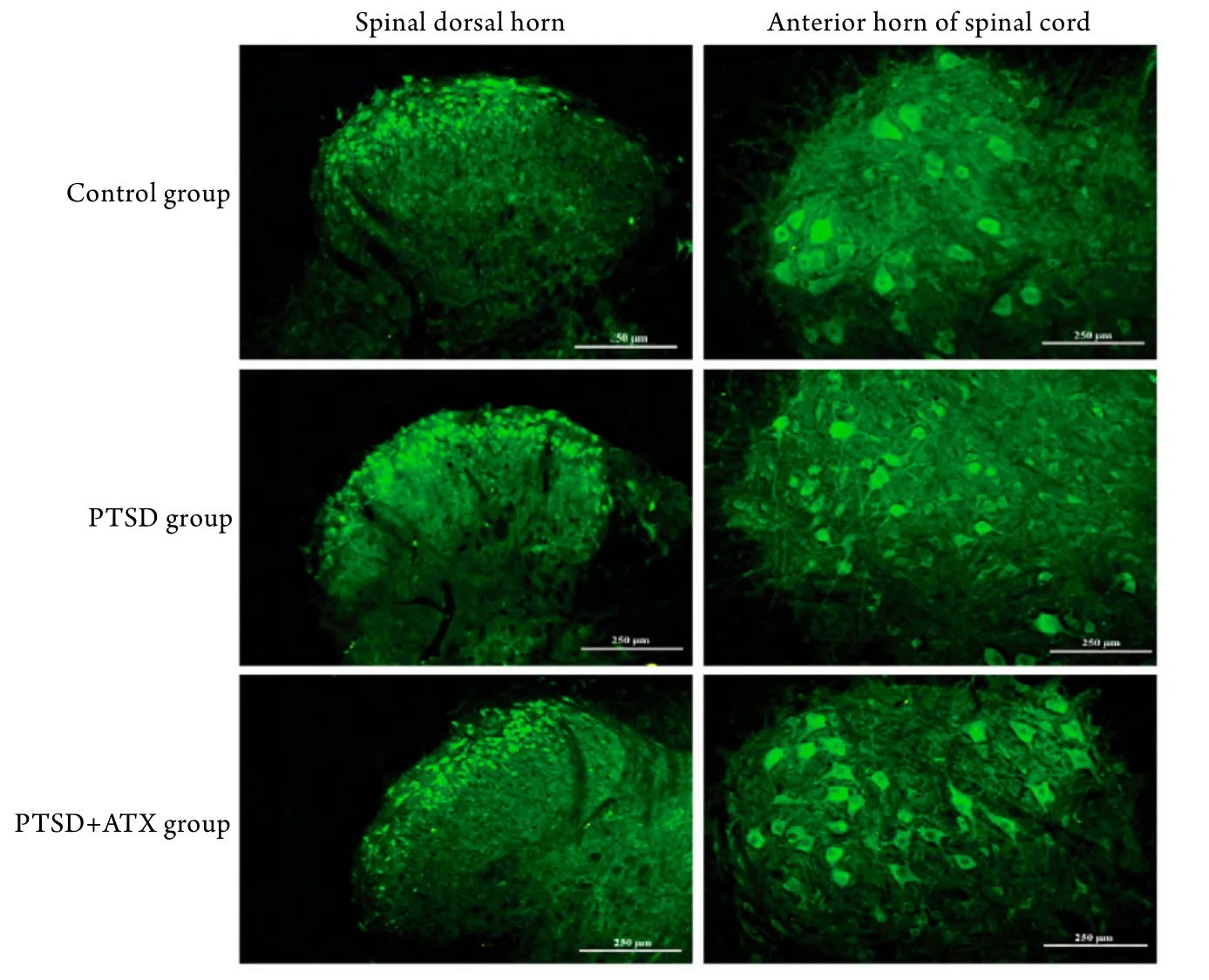
图1 脊髓组织HCN2表达的免疫组化检测(bar=250μm)Fig.1 HCN2 expression in spinal tissue (Immunohistochemistry, bar=250μm)
表1 不同CRD压力下大鼠内脏敏感性(AUCVMR值)比较(Tab.1 Comparison of visceral sensitivity (AUCVMR) of rats under different CRD
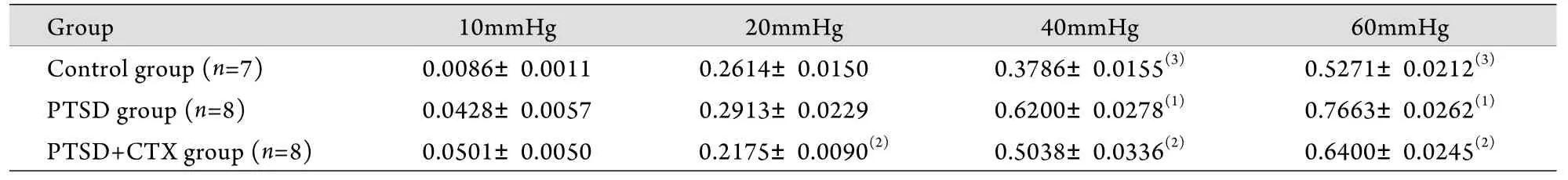
表1 不同CRD压力下大鼠内脏敏感性(AUCVMR值)比较(Tab.1 Comparison of visceral sensitivity (AUCVMR) of rats under different CRD
(1)P<0.001 compared with control group; (2)P<0.01 compared with PTSD group; (3)P<0.01 compared with PTSD+CTX group
Group 10mmHg 20mmHg 40mmHg 60mmHg Control group (n=7) 0.0086±0.0011 0.2614±0.0150 0.3786±0.0155(3) 0.5271±0.0212(3)PTSD group (n=8) 0.0428±0.0057 0.2913±0.0229 0.6200±0.0278(1) 0.7663±0.0262(1)PTSD+CTX group (n=8) 0.0501±0.0050 0.2175±0.0090(2) 0.5038±0.0336(2) 0.6400±0.0245(2)
3 讨 论
本研究采用SPS联合足底电击刺激成功建立了PTSD内脏高敏感大鼠模型,本模型在第7天时大鼠的内脏敏感性达到最高。本研究结果表明,在PTSD状态下大鼠脊髓HCN2表达显著上调,为HCN2参与应激相关的内脏高敏感性-中枢敏化的形成提供了形态学基础。另外,我们发现,在PTSD模型鼠脊髓前角运动神经元上也有大量HCN2的表达,可能与机械性痛敏和运动功能相关。GLT-1激动剂CTX可选择性诱发编码GLT-1的基因转录,同时可明显下调HCN2的表达,共同发挥抗内脏伤害性刺激的效应,提示在PTSD状态下HCN2-GLT-1可协同调控内脏高敏感性-痛觉敏化的形成。
既往研究表明,HCN2通道在炎性痛和神经病理性痛模型大鼠初级感觉神经元中的表达水平显著升高,并且在PGE2等因子的影响下,使脊髓背角神经元的兴奋性提高并导致神经元异常放电,而HCN2基因敲除则能明显减轻神经病理性/炎性疼痛模型小鼠的痛行为及痛敏状态,机械性诱发痛和热痛觉过敏明显减轻[14-15],提示HCN2通道在异常疼痛的发生及维持中发挥重要作用。本研究亦表明,在PTSD状态下HCN2在脊髓水平表达明显上调,其调控内脏高敏感性-中枢敏化形成的主要机制可能为:①通过HCN2介导Ih发挥作用。Ih是一种电压依赖性阳离子电流,它决定着神经元的静息膜电位高低及调控动作电位,还介导不同神经元神经冲动的节律性发放。有研究表明,神经损伤后,表达Ih的神经元比例明显升高,同时其Ih的电流激活曲线向去极化方向平移,半激活电位明显去极化,诱使Ih的电流活性增强[16],从而提示HCN2介导的Ih电流可能是调控PTSD状态下内脏高敏感性-中枢敏化形成的主要机制之一。②与GLT-1的协同调控作用。目前有关GLT-1-HCN2相互作用的确切分子机制尚不清楚,可能与GLT-1通过调节谷氨酸在突触前膜和突触后膜受体的利用率,双向调节突触内的谷氨酸水平,进而影响HCN2离子通道的表达有关。另外,既往研究亦证实[9-11],HCN2的阻断剂(ZD7288)可剂量依赖性抑制神经元谷氨酸释放及谷氨酸诱导的细胞内钙增加,从而抑制突触前Ca2+内流,调节突触前递质的释放。本研究表明,在PTSD状态下,采用CTX选择性诱发编码GLT-1的基因转录后,HCN2的表达明显下调,提示CTX可通过下调脊髓HCN2的表达参与调节内脏高敏感性-痛觉敏化的形成;另外,CTX还可通过上调GLT-1的表达减少谷氨酸累积[7],协同发挥抗伤害性内脏刺激作用。
总之,本研究表明,PTSD状态下脊髓水平HCN2通道表达发生了改变,采用CTX选择性诱发编码GLT-1的基因转录可影响HCN2的表达,提示PTSD状态下HCN2-GLT-1在脊髓水平具有协同调控内脏高敏感性-中枢敏化的作用。有效调控HCN2-GLT-1信号通路,可为防止或抑制PTSD及内脏高敏感性-痛觉敏化相关疾病的形成和发展提供新的治疗靶点。
[1]Javidi H, Yadollahie M. Post-traumatic stress disorder[J]. Int J Occup Environ Med, 2012, 3(1): 2-9.
[2]Kong XY, Xiao LN, Huang W, et al. Advances in epidemiological study of post-traumatic stress disorders among survivors of war trauma [J]. Med J Chin PLA, 2012, 37(2): 85-89. [孔祥毓, 肖立宁, 黄文, 等. 战后平民幸存者创伤后应激障碍的流行病学研究进展[J]. 解放军医学杂志, 2012, 37(2): 85-89.]
[3]Alschuler KN, Otis JD. Significant others' responses to pain in veterans with chronic pain and clinical levels of post-traumatic stress disorder symptomatology[J]. Eur J Pain, 2013, 17(2):245-254.
[4]Cohen H, Jotkowitz A, Buskila D, et al. Post-traumatic stress disorder and other co-morbidities in a sample population of patients with irritable bowel syndrome[J]. Eur J Intern Med,2006, 17(8): 567-571.
[5]Ludwig A, Zong X, Jeglitsch M, et al. A family of hyperpolarization-activated mammalian cation channels[J].Nature, 1998, 393(6685): 587-591.
[6]Moosmang S, Biel M, Hofmann F, et al. Differential distribution of four hyperpolarization-activated cation channels in mouse brain[J]. Biol Chem, 1999, 380(7-8): 975-980.
[7]Ramos KM, Lewis MT, Morgan KN, et al. Spinal upregulation of glutamate transporter GLT-1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders[J].Neuroscience, 2010, 169(4): 1888-1900.
[8]Yaster M, Guan X, Petralia RS, et al. Effect of inhibition of spinal cord glutamate transporters on inflammatory pain induced by formalin and complete Freund's adjuvant[J]. Anesthesiology,2011, 114(2): 412-423.
[9]Zheng M, Guo LJ, Xu XL, et al. ZD7288 inhibits the synaptic transmission in the pathway from perforant pathway fibers to CA3 region in rat hippocampus[J]. Yao Xue Xue Bao, 2006,41(6): 565-571.
[10]Chevaleyre V, Castillo PE. Assessing the role of Ihchannels in synaptic transmission and mossy fiber LTP[J]. Proc Natl Acad Sci USA, 2002, 99(14): 9538-9543.
[11]Yu X, Duan KL, Shang CF, et al. Calcium influx through hyperpolarization-activated cation channels (Ihchannels)contributes to activity-evoked neuronal secretion[J]. Proc Natl Acad Sci USA, 2004, 101(4): 1051-1056.
[12]He YQ, Chen Q, Ji L, et al. PKC gamma receptor mediates visceral nociception and hyperalgesia following exposure to PTSD-like stress in the spinal cord of rats[J]. Mol Pain, 2013,9(1): 9-35.
[13]Bradesi S, Golovatscka V, Ennes HS, et al. Role of astrocytes and altered regulation of spinal glutamatergic neurotransmission in stress-induced visceral hyperalgesia in rats[J]. Am J Physiol Gastrointest Liver Physiol, 2011, 301(3): G580-G589.
[14]Emery EC, Young GT, Berrocoso EM, et al. HCN2 ion channels play a central role in inflammatory and neuropathic pain[J].Science, 2011, 333(6048):1462-1466.
[15]Takasu K, Ono H, Tanabe M. Spinal hyperpolarization-activated cyclic nucleotide-gated cation channels at primary afferent terminals contribute to chronic pain[J]. Pain, 2010, 151(1):87-96.
[16]Cheng L, Sanguinetti MC. Niflumic acid alters gating of HCN2 pacemaker channels by interaction with the outer region of S4 voltage sensing domains[J]. Mol Pharmacol, 2009, 75(5): 1210-1221.
