淫羊藿通络丸薄层色谱鉴别方法的研究
2014-10-25吴世福
吴世福,王 红
(1.山东省医疗器械产品质量检测中心,山东济南250101;2.济宁市第一人民医院西院区,山东济宁271000)
淫羊藿通络丸由炙淫羊藿、桂枝、炒白术、茯苓、人参等7味中药组成,具有温补脾肾、祛湿通络的功效,用于糖尿病周围神经病变之脾肾阳虚、湿瘀阻络证。本文通过对样品处理方法、展开剂的考察,建立了专属性强、简便可行的薄层色谱鉴别方法,可有效地控制产品质量,保证临床用药安全有效。
1 仪器与试药
1.1 仪器 十万分之一电子分析天平(密特勒-托利多仪器有限公司);万分之一电子分析天平(北京赛多利斯天平有限公司);数显恒温水浴锅(金坊市医疗仪器厂);智能电热恒温鼓风干燥箱(上海琅轩实验设备有限公司);超声波清洗器(上海科导超声波有限公司);硅胶G(青岛海洋化工厂);定量毛细管(日本岛津),MERCK薄层层析板(批号:OB678541)。
1.2 试药 淫羊藿苷(批号:110737-200415)、人参皂苷Rb1对照品(批号:110704-200921)、人参皂苷Re对照品(批号:110754-200822)、人参皂苷Rg1对照品(批号:110703-201128)、川芎对照药材(批号:120918-201110)、芍药苷对照品(批号:110736-201136)、白术对照药材(批号:120925-201109)桂枝对照药材(批号:121191-201103)、人参对照药材(批号:120917-200609),均由中国药品生物制品检定所提供;淫羊藿通络丸(批号为120801、120403、120902,由济宁市中医院提供);阴性样品(自制):按处方工艺分别制备不同药材相应的阴性样品;试剂均为分析纯。
2 方法与结果
2.1 炙淫羊藿 取本品14 g,研细,加乙醇80 mL,超声处理30 min,滤过,滤液蒸干,残渣加水20 mL使溶解,用乙酸乙酯提取3次,每次20 mL,水溶液备用;合并乙酸乙酯提取液,蒸干,残渣加甲醇2 mL溶解,作为供试品溶液。取不含炙淫羊藿的阴性样品,同法制备阴性样品溶液。另取淫羊藿苷对照品,加甲醇制成每1 mL含0.2 mg的溶液,作为对照品溶液。照薄层色谱法[《中国药典》2010年版(一部)附录Ⅵ B]试验,吸取上述三种溶液各6 μL,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶 G薄层板,以乙酸乙酯 - 丁酮 - 甲酸 - 水(10∶1∶1∶1)为展开剂,展开,取出,晾干,喷以三氯化铝试液,加热至斑点清晰,置紫外光灯(365 nm)下检视供试品色谱中,在对照品色谱相应的位置上,显相同颜色的荧光斑点。阴性样品无干扰,见图1。

图1 淫羊藿薄层色谱图
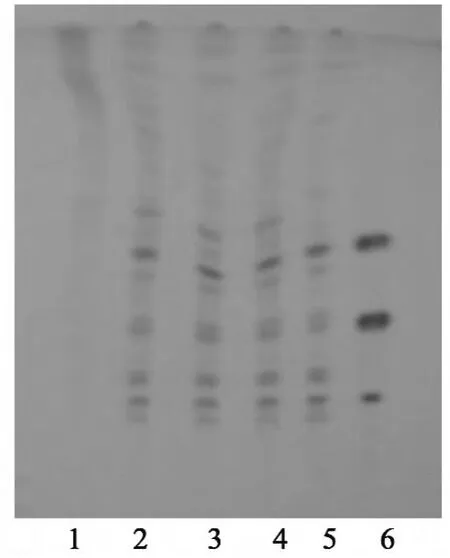
图2 人参薄层色谱图
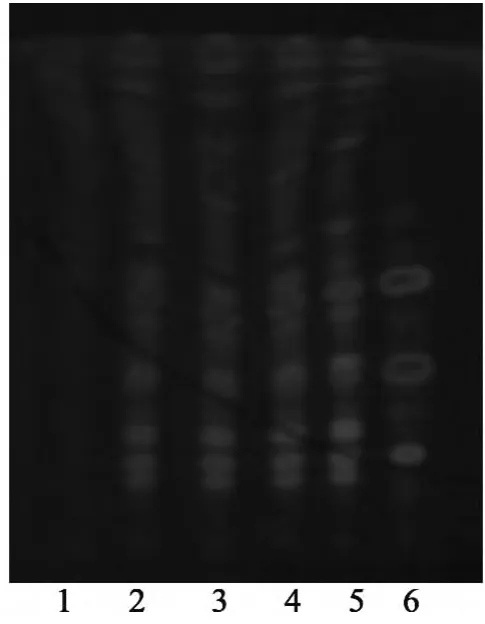
图3 人参薄层色谱图
2.2 人参 取“2.1”项下的水溶液,加水饱和正丁醇振摇提取3次,每次40 mL,合并正丁醇液,依次用浓氨试液25 mL、正丁醇饱和的水25 mL各洗涤2次,分取正丁醇液,蒸干,残渣加水10 mL使溶解,通过D101大孔树脂柱(内径为1.5 cm,柱高为12 cm),依次用水50 mL、20%乙醇50 mL洗脱,弃去洗脱液,继用80%乙醇80 mL洗脱,收集洗脱液,蒸干,残渣加甲醇1 mL溶解,作为供试品溶液。取不含人参的阴性样品,同法制备阴性样品溶液。另取人参对照药材1 g,加乙醇30 mL,超声处理30 min,滤过,残渣加水20 mL溶解,用乙酸乙酯提取3次,每次20 mL,弃去乙酸乙酯液,自“水溶液加水饱和正丁醇振摇提取”起,同法制成对照药材溶液。再取人参皂苷Rb1对照品、人参皂苷Re对照品及人参皂苷Rg1对照品,加甲醇制成每1 mL各含2 mg的混合溶液,作为对照品溶液。照薄层色谱法[《中国药典》2010年版(一部)附录ⅥB]试验,吸取供试品溶液、阴性样品溶液和对照药材溶液各10 μL、对照品溶液4 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(65∶35∶10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品色谱相应位置上,分别显相同颜色的斑点;紫外光灯(365 nm)下检视,显相同颜色的荧光斑点。阴性样品无干扰,见图2~3。
2.3 川芎 取本品粉末14 g,加乙醚40 mL,超声处理30 min,滤过,药渣备用;滤液挥干,残渣加乙酸乙酯2 mL使溶解,作为供试品溶液。取不含川芎的阴性样品,同法制备阴性样品溶液。另取川芎对照药材1 g,同法制成对照药材溶液。照薄层色谱法[《中国药典》2010年版(一部)附录Ⅵ B)试验,吸取上述三种溶液各4 μL,分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯(9∶1)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品色谱图中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。阴性样品无干扰,见图4。
2.4 炒白芍 取“2.3”项下的药渣,加甲醇50 mL,超声处理30 min,滤过,滤液蒸干,残渣加水15 mL使溶解,加水饱和正丁醇振摇提取3次,每次40 mL,合并正丁醇液,蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。取不含炒白芍的阴性样品,同法制备阴性样品溶液。另取芍药苷对照品,加甲醇制成每1 mL含2 mg的溶液,作为对照品溶液。照薄层色谱法[《中国药典》2010年版(一部)附录Ⅵ B]试验,吸取供试品溶液、阴性样品溶液各6 μL及对照品溶液4 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-乙酸乙酯-浓氨试液(50∶20∶10∶2.5)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,热风吹至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。阴性样品无干扰,见图5。

图4 川芎薄层色谱图

图5 炒白芍薄层色谱图
2.5 炒白术 取本品粉末14 g,置烧瓶中,加水200 mL,照挥发油测定法[《中国药典》2010年版(一部)附录ⅩD]试验,自冷凝管上端加石油醚(60~90℃)1.5 mL,加热至沸并保持微沸2 h,分取石油醚液,作为供试品溶液。取不含炒白术的阴性样品,同法制备阴性样品溶液。另取白术对照药材2 g,同法制备对照药材溶液。照薄层色谱法[《中国药典》2010年版(一部)附录ⅥB]试验,吸取上述三种溶液各4~6 μL,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,并应显有一桃红色主斑点。阴性样品无干扰,见图6。

图6 炒白术薄层色谱图
2.6 桂枝 取桂枝对照药材2 g,置烧瓶中,加水200 mL,照挥发油测定法[《中国药典》2010年版(一部)附录ⅩD]试验,自冷凝管上端加石油醚(60~90℃)1.5 mL,加热至沸并保持微沸2 h,分取石油醚液,作为对照药材溶液。取不含桂枝的阴性样品,同法制备阴性样品溶液。照薄层色谱法[《中国药典》2010年版(一部)附录ⅥB]试验,吸取阴性样品溶液、“2.5”项下的供试品溶液4~8 μL及上述对照药材溶液4 μL,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-乙酸乙酯(17∶3)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。置紫外光灯(365 nm)下检视,显相同颜色的荧光斑点。阴性样品无干扰,见图7~8。
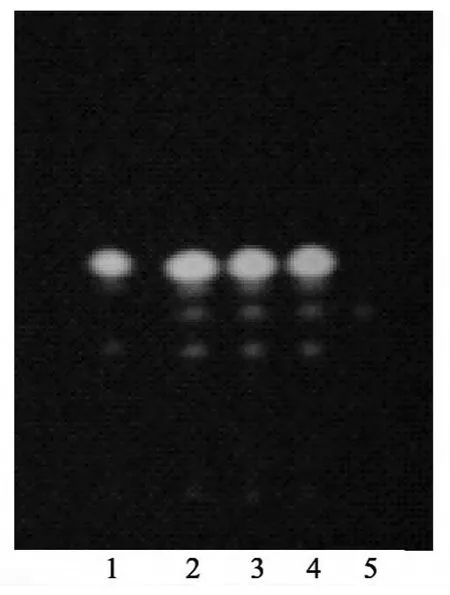
图7 桂枝薄层色谱图
3 讨论
3.1 本品为中药复方制剂,参考有关文献[1~6],采用薄层色谱鉴别方法对方中的主要药味均进行了鉴别,并进行了阴性对照试验,结果其方法简便且专属性强,可有效控制本品的质量。

图8 桂枝薄层色谱图
3.2 提取方法的选择 分别以乙醚、正己烷为溶剂,采用超声波提取法及提取挥发油方法制备供试品溶液,对白术和桂枝进行薄层鉴别,结果以提取挥发油法效果较好,薄层色谱斑点清晰,阴性样品无干扰。
[1]许闽,王淑美,冯素香,等.补肾壮阳胶囊的定性研究[J].河南中医学院学报,2007,22(133):33 -34.
[2]付玮辰,白海玉,韩德强.参金益气丸质量标准的研究[J].黑龙江医药,2010,23(4):55 -56.
[3]国家药典委员会.中华人民共和国药典2010年版(一部)[S].北京:中国医药科技出版社,2010.
[4]杜婷婷,陈智.淫羊藿有效成分淫羊藿苷的现代研究进展[J].齐鲁药事,2012,31(9):533 -535.
[5]李凯,徐秀珍.人参健脾丸的薄层色谱鉴别研究[J].宁夏医科大学学报,2011,33(8):105 -106.
[6]高辉,马小军,林永强,等.健脑补肾丸定性定量方法的研究[J].时珍国医国药,2012,23(1):182-184.
