尼可地尔对阿霉素损伤大鼠心肌的保护作用
2014-10-17刘梦林王梦龙
刘梦林 万 军 王梦龙 石 磊 冯 颖
武汉大学人民医院,湖北武汉 430000
阿霉素是蒽环类抗肿瘤药物。自从1970年用于临床治疗肿瘤以来,被认为是治疗肿瘤的最有效的药物之一。但是,1973年,Lefrak首次报道了阿霉素对全身各脏器及组织均有毒性作用,其中对心脏的毒性作用尤为明显。因此,阿霉素在临床上的应用受到了限制[1]。一直以来,关于阿霉素心脏毒性的机制存在争议,并未能得到一个完整的阐述。其中阐述得比较多的机制主要有:①阿霉素产生的自由基诱导心肌细胞氧化应激[2],从而导致心肌细胞损伤甚至死亡。②阿霉素产生的氧自由基通过促进内质网打开Rya(ryanodine receptor)和损伤钙离子清楚系统,释放钙离子,增加细胞内的钙水平[3-4],从而促进心肌细胞的凋亡。③近来许多研究发现,阿霉素引起心肌损伤与阿霉素促进心肌细胞自噬有关[5-6]。目前,许多研究利用阿霉素对心肌的毒性作用,将其用于制造扩张型心肌病的模型。
ATP敏感钾通道(KATP)是是电压非依赖性的、配体门控通道,由Noma等[7]于1983年首先在豚鼠的心肌细胞上发现。随后多个研究发现,ATP敏感钾通道还存在于人体多种器官及组织中,例如血管平滑肌细胞、骨骼肌细胞、神经元、胰腺细胞等。研究表明,它是由四个内向整流钾通道(Kir6.x家族)亚单位和 4个调节性磺脲类受体(SUR)组成的八聚体[8]。Kir6.x家族已发现两组成员:Kir6.1和 Kir6.2,SUR也发现 2种:SURl和 SUR2,SUR2又有 2种不同的剪接体形式即SUR2A和SUR2B。KATP通道的活性受ATP/ADP比例的调节,当ATP/ADP比例下降时,KATP通道被激活。近年来,许多研究发现KATP通道的激活能对心脏产生保护作用,例如,KATP通道的激活是缺血预适应的重要环节。它的开放剂(尼可地尔、二氮嗪)目前已应用于临床治疗高血压、心绞痛等疾病[9-10]。
本实验拟在前人的研究基础上,用阿霉素制造心肌损伤模型,然后用尼可地尔进行干预,探讨尼可地尔对阿霉素损伤心肌是否能产生保护作用。
1 材料与方法
1.1 实验动物
45只6~7周健康雄性SD大鼠(北京华阜康生物科技股份有限公司提供,动物使用许可证号:SCXK(京)2009-0004),体重 150~180 g。 随机分为 3 组,每组各15只。三组大鼠的体重、年龄差异均无统计学意义(P > 0.05),具有可比性。
1.2 实验试剂与仪器
尼可地尔(购置Sigma公司,货号为N3539);阿霉素(购置武汉大学人民医院试剂科,产品批号为2QL 0051);生理盐水;超声仪(美国 Acuson Sequoia型彩超仪,心脏探头7V3,频率为6.0 MHz)。
1.3 实验方法
1.3.1 分组 45只雄性SD大鼠用随机数字表随机分为3组(每组各15只)。分别为对照组(A组)、阿霉素模型组(B组)、尼可地尔+阿霉素组(C组)。
1.3.2 造模 B、C组大鼠按2.5 mg/kg的量腹腔注射阿霉素,A组大鼠按2.5 mg/kg的量腹腔注射生理盐水,1周1次,共持续6周。停药后观察2周,第8周开始C组大鼠按每次3 mg/kg的量用灌胃法灌入尼可地尔,1 次/d,持续 30 d。
1.3.3 超声检测 造模结束后,给各组大鼠称重,并根据大鼠的体重按3.5 mg/100 g的剂量用1%的戊巴比妥钠(双蒸水稀释)麻醉,然后用超声仪器进行心脏功能检测,检测的指标包括舒张末内径(LVEDD)、左室收缩末内径(LVESD)、左室射血分数(LVEF)、收缩末期容积(ESV)、舒张末期容积(EVD)及左室短轴收缩率(FS%)。
1.3.4 心肌形态学检测 超声测定完毕后,将大鼠按每3.5 mg/100 g的剂量腹腔注射1%的戊巴比妥钠进行深度麻醉,打开胸腔,暴露心脏,游离大鼠的心脏,并将其放入10%KCl液中清洗掉血液,在干冰上切取其中部分心脏组织(横向切)放入10%的福尔马林中固定,然后石蜡包埋、切片、天狼星红染色、显微镜下观察并拍片,最后用IPP 6.0软件进行分析,计算心肌组织的胶原分数[11-12]。计算公式为:胶原容积分数=胶原面积/(整个视野的总面积-空白面积)×100%。
1.4 统计学方法
采用统计软件SPSS 18.0对实验数据进行分析,正态分布计量资料数据以均数±标准差(±s)表示,多组均数比较用方差分析,组间两组比较行LSD-t检验。以P<0.05为差异有统计学意义。
2 结果
2.1 大鼠一般情况
造模期间,A组大鼠全部存活,B组存活10只,C组存活11只。另B、C两组均有部分大鼠出现活动减少、腹泻、血性腹水、胸腔积液、体重增长速度减慢等情况。造模结束时,B组和C组大鼠平均体重小于A组(P<0.01),而且 B组大鼠平均体重小于 C组(P<0.05)。 见表1。
2.2 超声结果
与A组比较,B组大鼠的心脏功能明显受损,LVEDS、ESV 明显增大(P < 0.01),LVEF、FS%明显降低(P<0.01);而与 B 组比较,C 组 LVEDS、ESV 明显减小(P < 0.01),LVEF、FS%明显增高(P < 0.01)。 见表1。
表1 超声检查结果及体重(±s)
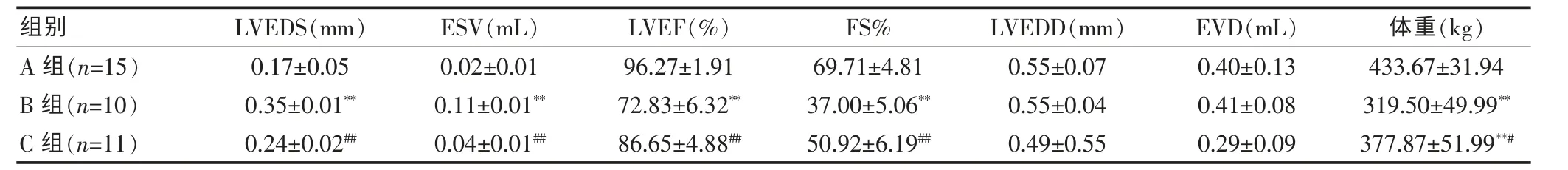
表1 超声检查结果及体重(±s)
注:与 A 组比较,**P < 0.01;与 B 组比较,#P < 0.05,##P < 0.01;LVEDS:左室收缩末内径;ESV:收缩末期容积;LVEF:左室射血分数;LVEDD:左室舒张末内径;EVD:舒张末期容积;FS%:室短轴收缩率
组别 LVEDS(mm) ESV(mL) LVEF(%) FS% LVEDD(mm) EVD(mL) 体重(kg)A 组(n=15)0.17±0.050.02±0.0196.27±1.9169.71±4.810.55±0.070.40±0.13433.67±31.94 B 组(n=10)0.35±0.01**0.11±0.01**72.83±6.32**37.00±5.06**0.55±0.040.41±0.08319.50±49.99**C 组(n=11)0.24±0.02##0.04±0.01##86.65±4.88##50.92±6.19##0.49±0.550.29±0.09377.87±51.99**#
2.3 心肌形态学检测结果
超声检测结束后,取部分心肌组织行天狼星红染色,在光镜下可见经阿霉素处理后B、C组大鼠心肌组织均有不同程度纤维化,且均明显高于A组,而给予尼可地尔灌胃后的C组大鼠心肌组织纤维化程度低于 B组(图1,天狼星红染色,400×)。 另外,B组心肌组织胶原容积分数为 (3.79±0.98)%明显比 A组[(1.58±0.41)%]高,两者比较差异有高度统计学意义(P<0.01),C组心肌组织胶原容积分数为 (2.87±0.93)%,与B组比较差异无统计学意义(P>0.05)。

图1 各组心肌组织光镜下图片(天狼星红染色,400×)
3 讨论
尼可地尔属硝酸酯类化合物,是由N-2(2-羟乙基)烟酰胺维生素和有机硝酸酯的部分结构连接而成的化合物。它是一种ATP敏感性钾通道开放剂,能舒张动脉血管,同时由于它的结构中具有硝酸酯基,也能舒张静脉血管。它兼有舒张两种血管的作用,所以是用于治疗心绞痛的新型药物。更值得注意的是,越来越多的试验结果提示:钾通道激活剂也通过增强能保护心脏不发生局部缺血的正常生理过程而发挥直接的细胞保护作用。主要机制可能为:①维持细胞的能量代谢:mitoKATP通道开放后,K+进入线粒体,线粒体内渗透压升高,引起基质肿胀,并激活电子传递链,促进线粒体呼吸,使ATP生成增多,为细胞提高更多的能量[13];另外,可通过保护的结构和功能,使缺血缺氧期细胞内高能磷酸化合物丢失减少。②减少Ca2+超载:KATP通道开放后,可减少Ca2+内流,从而减轻线粒体内Ca2+超载[14]。③减少细胞凋亡:KATP通道开放后,抑制线粒体渗透性转换孔(MPTP)开放,从而抑制其下游凋亡蛋白的级联反应,起到抗细胞凋亡的作用。④抗自由基的生成:KATP通道开放逆转钙超载,稳定细胞膜,保护细胞膜的完整功能,减少氧自由基的产生。
本实验通过采用阿霉素制造阿霉素损伤大鼠心肌的模型,模拟人类的扩张型心肌病,然后用尼可地尔进行干预,探讨尼可地尔对阿霉素损伤大鼠心肌是否有保护作用。本实验的结果提示阿霉素模型组大鼠的心脏功能明显差于正常对照组,且心肌组织出现明显的纤维化,说明阿霉素可致大鼠心脏功能下降和心肌纤维化。采用尼可地尔进行干预后,大鼠心脏功能明显改善,且心肌纤维化程度减轻,说明尼可地尔对阿霉素损伤心肌有保护作用,这跟众多研究所证实的尼可地尔对缺血再灌注心脏有保护作用的结果一致。另外,尼可地尔干预组心肌纤维化程度比阿霉素组轻,而心肌纤维化是心肌氧化损伤或坏死、凋亡后的一种病理反应,说明尼可地尔能抗氧化损伤及细胞凋亡,这与之前的研究结果一致。但阿霉素模型组与尼可地尔干预组心肌胶原容积分数之间的差异并无统计学意义,可能的原因有:①本实验采用的样本量不够大,加上组内课题差异较大,故有待增加样本量行进一步研究。②可能尼可地尔的剂量比较小,抗心肌纤维化的作用还不够明显。③心肌纤维化不单单是心肌氧化损伤或坏死、凋亡后的一种病理反应,它还与其他多种因素有关,例如肾素-血管紧张素-醛固酮系统(RAAS)、免疫系统和多种细胞因子等[15],可能尼可地尔与这些因素相互作用后,削弱了它抗心肌纤维化的作用。这些均有待行进一步研究,以期望尼可地尔将来能用于临床心肌病患者的治疗中。
[1]Amie JD.The role of autophagy in doxorubicin-induced cardiotoxicity[J].Life Sciences,2013,93(24):913-916.
[2]Zhang S,Liu X,Bawa-Khalfe T,et al.Identification of the molecular basis of doxorubicin-induced cardiotoxicity[J].Nat Med,2012,18:1639-1642.
[3]Camello-Almaraz C,Gomez-Piilla PJ,Pozo MJ, et al.Mitochondrial reactive oxygen species and Ca2+signaling[J].Am J Physiol Cell Physiol,2006,291(5):1082-1088.
[4]Kim SY,Kim SJ,Kim BJ,et al.Doxorubicin-induced reactive oxygen species Generation and intracellular Ca2+increase are reciprocally modulated in rat Cardiomyocytes[J].Exp Mol Med,2012,38(5):535-545.
[5]Smuder AJ,Kavazis AN,Min K,et al.Doxorubicin-induced markers of myocardial autophagic signaling in sedentary and exercise trained animals [J].J Appl Physiol,2013,115(176):85.
[6]Sishi BJ,Loos B,Van Rooyen J,et al.Autophagy upregulation promotes survival and attenuates doxorubicin-induced cardiotoxicity[J].Biochem Pharmacol,2013,85:124-134.
[7]Noma A,Shibasaki T.Membrane current through adenosinetriphosphate-regulated potassium channels in guinea-pig ventricular cells[J].J Physiol,1985,363:463-480.
[8]Eirini K,Li B,Michael JR,et al.KATP channels as molecular sensors of cellular metabolism [J].Nature,2013,440(7083):470-476.
[9]徐林,张登文,张传汉,等.心肌细胞膜ATP敏感性钾离子通道——从基础到临床[J].医学分子生物学杂志,2013,10(5):299-303.
[10]Hsi CY,Fang SY,Chen YZ,et al.Cardiovascular protection of activating KATP channel during ischemia-reperfusion acidosis[J].Shock,2012,37(6):653-658.
[11]Ding WY,Liu L,Wang ZH,et al.FP-receptor gene silencing ameliorates myocardial fibrosis and protects from diabetic cardiomyopathy[J].Journal of Molecular Medicine,2012,(56):596-607.
[12]Querejeta R,Varo N,Lopez B,et al.Serum carboxy-terminal propertide of procollagen typeⅠis a marker of myocardial fibrosis in hypertensive heart disease[J].Circulation,2000,101:1729-1735.
[13]Swyers T,Redford D,Larson DF.Volatile anesthetic-induced preconditioning[J].Perfusion,2014,29(1):10-15.
[14]Calderone V,Testail L,Martelli A,et al.Anti-ischemie properties of a new spirocyclic benzopyran activator of the mito-KATP channel[J].Biochem Pharmacol,2010,79(1):39-47.
[15]Kuwahara F,Kai H,Tokuda K,et al.Transforming growth factor-beta function Bloking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats[J].Circulation,2002,106:130-135.
