5-甲基胞嘧啶锁核酸的合成新方法*
2014-08-30顾问,陈贝,葛敏
顾 问,陈 贝,葛 敏
(南京工业大学 生物与制药工程学院 材料化学工程国家重点实验室,江苏 南京 210009)
·研究简报·
5-甲基胞嘧啶锁核酸的合成新方法*
顾 问,陈 贝,葛 敏
(南京工业大学 生物与制药工程学院 材料化学工程国家重点实验室,江苏 南京 210009)
设计了一种新的合成5-甲基胞嘧啶锁核酸{(1R,3R,4R,7S)-3-[(5-甲基-4-N-苯甲酰基胞嘧啶-1-基)-7-(2-氰基乙氧基)-(N,N-二异丙基)膦氧代]-1-(4,4′-二甲氧基三苯代甲基氧基甲基)-2,5-二氧杂二环-[2.2.1]庚烷(1)}的方法。以3-苄氧基-4-C-羟甲基-1,2-O-异亚丙基-α-D-呋喃核糖为起始原料,经取代、水解等12步反应合成了1,总收率21.0%,其结构经1H NMR和MS确证。
锁核酸;呋喃核糖;胞嘧啶;合成
锁核酸(LNA)是一种经过修饰的核糖核酸类寡核苷酸衍生物,结构中腺嘌呤、胞嘧啶、鸟嘌呤、胸腺嘧啶、尿嘧啶和5-甲基胞嘧啶六种碱基核糖的2′-O,4′-C位通过缩水作用形成刚性结构[1]。与其他寡核苷酸类似物相比,LNA具有很好的热稳定性、水溶性及体内无毒副作用等优点,广泛应用于基因诊断、基因治疗、PCR等技术中[2-3]。LNA中六种碱基磷酸盐核糖是其重要组成部分,其中β型5-甲基胞嘧啶锁核酸单体的合成方法鲜有报道。Alexei A Koshkin小组[4]以3-苄氧基-4-C-羟甲基-1,2-O-异亚丙基-α-D-呋喃核糖(2)[5]为原料,经取代、缩合、水解等8步反应先合成胸腺嘧啶呋喃核糖中间体,再经1,2,4-三氮唑取代、水解将胸腺嘧啶转换为5-甲基胞嘧啶锁核酸{(1R,3R,4R,7S)-3-[(5-甲基-4-N-苯甲酰基胞嘧啶-1-基)-7-(2-氰基乙氧基)-(N,N-二异丙基)膦氧代]-1-(4,4′-二甲氧基三苯代甲基氧基甲基)-2,5-二氧杂二环-[2.2.1]庚烷(1)},共经15步反应,总收率7.50%。该合成方法路线较长,收率较低。
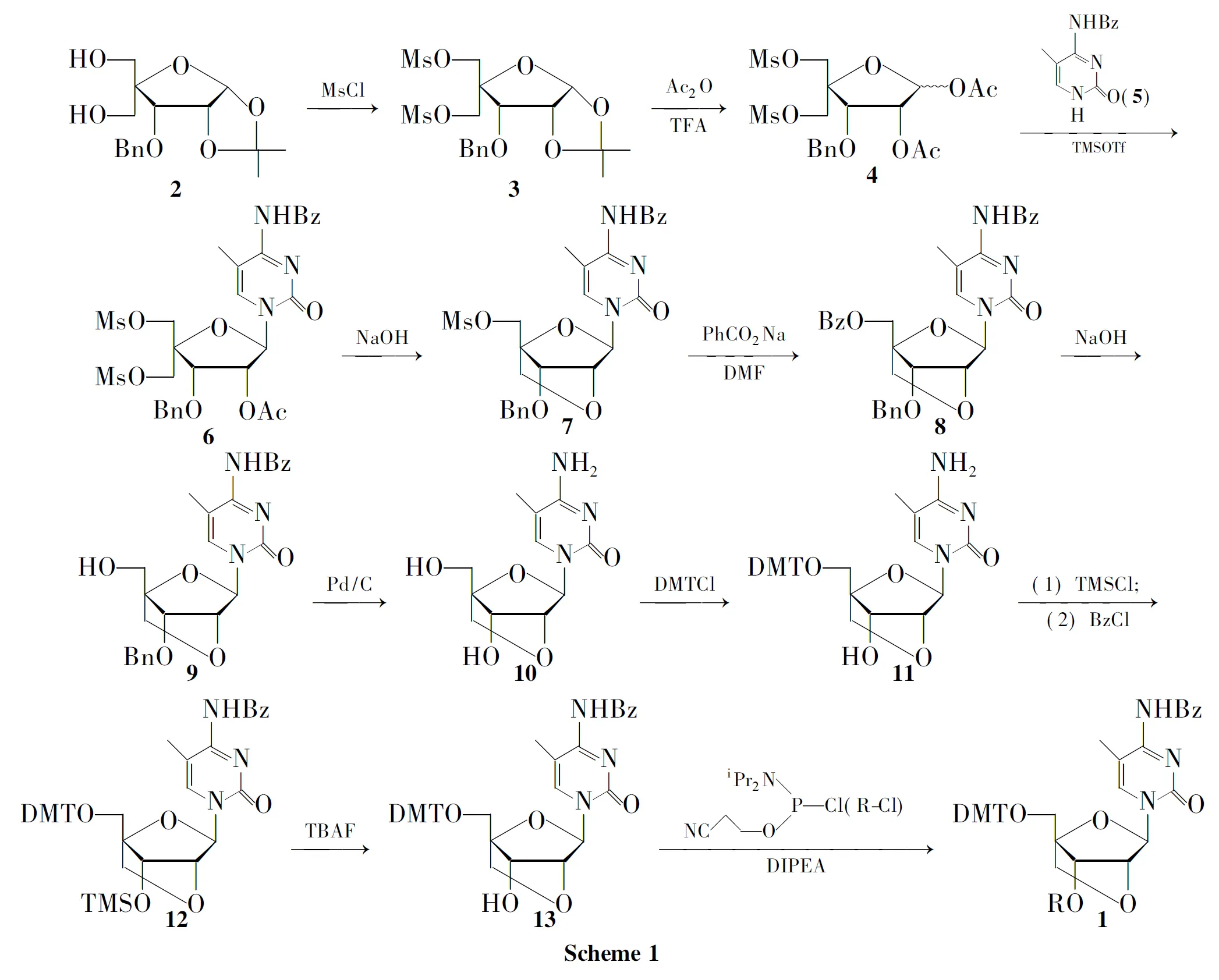
本文对1的合成进行了探索和研究。对文献[4]方法加以改进,设计了一种新的合成1的方法。以2为起始原料,经取代、水解等12步反应合成了1(Scheme 1),总收率21.0%,其结构经1H NMR和MS确证。
该方法采用三氟甲磺酸三甲基硅酯(TMSOTf)催化缩合[6]将4-N-苯甲酰氨基-5-甲基胞嘧啶(5)引入到目标化合物中,将反应路线的线性步数由15步减少至12步,同时改进了合成工艺,总收率从7.50%提高至21.0%,并使该路线更适合于合成其它不同碱基取代的核糖衍生物。
1 实验部分
1.1 仪器与试剂
Bruker DRX300/500MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent 1100型LC/MS液相色谱质谱联用仪。
所用试剂均为分析纯。
1.2 合成
(1)3-O-苯甲基-4-C-甲基磺氧基甲基-5-O-甲磺酰-1,2-O-异亚丙基-α-D-呋喃核糖(3)的合成[5]
在反应瓶中加入215.0g(48.6mmol)和吡啶72.0mL,冰水浴冷却,搅拌下缓慢滴加甲磺酰氯(MsCl)14.9g(130mmol),滴毕,于室温反应1h(TLC跟踪)。减压蒸除溶剂,加水(300mL)稀释,用二氯甲烷(2×100mL)萃取,合并萃取液,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,旋蒸脱溶得黄色油状液体318.6g,收率82%;1H NMRδ:7.26~7.36(m,5H),5.79(d,J=7.3Hz,1H),4.75~4.89(m,2H),4.63~4.66(m,1H),4.55~4.58(s,1H),4.30~4.43(m,2H),4.13~4.20(m,2H),3.08(s,3H),2.98(s,3H),1.68(s,3H),1.34(s,3H)。
(2)1,2-二-O-乙酰基-3-O-苯甲基-4-C-甲基磺氧基甲基-5-O-甲磺酰-D-呋喃核糖(4)的合成
在反应瓶中加入318.0g(39.0mmol),冰水浴冷却,搅拌下缓慢滴加80%三氟乙酸(TFA)溶液(113mL),滴毕,于室温反应1h(TLC跟踪)。用饱和碳酸钠溶液调pH至7~8,用二氯甲烷(2×100mL)萃取,合并萃取液,用饱和氯化钠溶液洗涤,无水硫酸钠干燥得油状液体中间体M。加入乙酸酐11.9g (0.117mol)和吡啶39mL,搅拌下于室温反应16h(TLC跟踪)。旋蒸除溶,残留物加水200mL,用乙酸乙酯(2×80mL)萃取,合并萃取液,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,浓缩得棕色油状液体418.7g,收率94%;1H NMRδ:7.26~7.37(m,5H),5.38(s,1H),4.59~4.63(m,1H),4.47~4.53(m,2H),4.39~4.42(m,2H),4.27~4.33(m,2H),4.15~4.25(m,1H),3.00~3.02(m,6H),2.14(s,3H),2.08(s,3H)。
由于 PCS9000数据库中设备类表的关键字为自动生成,如果两个系统同时修改模型数据就有可能会出现同一关键字所对应的设备不同的情况,从而造成两个系统模型数据的混乱,因此维护操作只能在其中一个系统进行。同时,当网络不通或者备用系统服务器发生故障时,主系统题,应将修改信息保存在本地机器上,等故障排除后,再将保存的信息发送给备用系统,用系统数据服务器执行修改操作,从而实现两系统之间图形和数据库的同步
(3)1-(2-O-乙酰基-3-O-苯甲基-4-C-甲基磺氧基甲基-β-D-呋喃核糖基)-4-N-苯甲酰氨基-5-甲基胞嘧啶(6)的合成[6]
在反应瓶中加入480.0g(157mmol),乙腈1.65L,554.0g(235mmol),N,O-双(三甲硅基)乙酰胺144mL(235mmol),搅拌下回流反应至反应液变澄清。冰水浴冷却,滴加TMSOTf 61.0mL(313mmol),滴毕,回流反应过夜(TLC跟踪)。减压蒸除溶剂,残留物加水(1.5L)稀释,用二氯甲烷(2×500mL)萃取,合并萃取液,依次用碳酸氢钠溶液、饱和氯化钠溶液洗涤,无水硫酸钠干燥,浓缩得黄色固体6104.0g,收率97.6%;1H NMRδ:13.21(s,1H),8.29(t,J=8.1Hz,2H),7.23~ 7.56(m,9H),5,73(s,1H),5.56~5.74(m,1H),4.69(d,J=6.6Hz,1H),4.34~4.63(m,6H),3.11(s,3H),3.00(d,J=7.8Hz,3H),2.17(s,3H),2.11(s,3H),2.04(s,3H);ESI-MSm/z: 680.6{[M+H]+}。
(4)(1S,3R,4R,7S)-7-苄氧基-1-甲基磺氧基甲基-3-(5-甲基-4-N-苯甲酰基胞嘧啶-1-基)-2,5-二氧杂二环-[2.2.1]庚烷(7)合成
在反应瓶中加入660.0g(88.6mmol),1,4-二氧六环750mL和1mol·L-1氢氧化钠溶液(750mL),搅拌下于室温反应1h(TLC跟踪)。用二氯甲烷(3×500mL)萃取,合并萃取液,用无水硫酸钠干燥,浓缩得黄色固体747.0g,收率99%;1H NMRδ:13.35(s,1H),8.31(t,J=7.2Hz,2H),7.26~7.59(m,9H),5.71(s,1H),4.52~4.75(m,8H),3.09(s,3H),2.12(s,3H);ESI-MSm/z: 542.5{[M+H]+}。
(5)(1S,3R,4R,7S)-7-苄氧基-1-苯甲酰氧基甲基-3-(5-甲基-4-N-苯甲酰基胞嘧啶-1-基)-2,5-二氧杂二环-[2.2.1]庚烷(8)的合成
在反应瓶中加入747.0g(88.5mmol),DMF 441mL,苯甲酸钠 23.0g(160mmol),搅拌下于90℃反应过夜(TLC跟踪)。加水(1.2L)稀释,用二氯甲烷(3×500mL)萃取,合并萃取液,用无水硫酸钠干燥,浓缩,残留物加甲醇1L(析出固体),抽滤,滤饼真空干燥得白色固体831g,收率62%;1H NMRδ:8.28(t,J=8.5Hz,2H),7.97(t,J=9.0Hz,2H),7.61~7.65(m,2H),7.44~7.55(m,5H),7.25~7.49(m,5H),5.62(s,1H),4.85(d,J=12.5Hz,1H),4.71(d,J=12.5Hz,2H ),4.52(d,J=12.5Hz,2H),4.19(d,J=12.0Hz,1H),3.98(d,J=8.0Hz,1H),3.98(s,1H),1.80(s,3H);ESI-MSm/z: 568.6{[M+H]+}。
(6)(1S,3R,4R,7S)-7-苄氧基-1-羟甲基-3-(5-甲基-4-N-苯甲酰基胞嘧啶-1-基)-2,5-二氧杂二环-[2.2.1]庚烷(9)的合成
在反应瓶中依次加入868.0g(119mmol),1,4-二氧六环(1L),1mol·L-1NaOH溶液1L,搅拌下于室温反应0.5h(TLC跟踪)。用2mol·L-1盐酸调pH至6,用二氯甲烷(3×500mL)萃取,合并萃取液,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩,残余物用混合溶剂[A=V(EA)∶V(PE)=5∶95]500mL分散,抽滤,滤饼真空干燥得白色固体952.0g,收率94.5%;1H NMRδ:8.30(t,J=7.5Hz,2H),7.70(s,1H),7.46(t,J=14.5Hz,1H),7.25~7.55(m,7H),5.70(s,1H),4.66(d,J=12.0Hz,1H),4.58(s,1H),4.54(d,J=12.0Hz,1H),3.95~4.05(m,4H),3.83(d,J=8Hz,1H),2.15(s,3H);ESI-MSm/z: 464.5{[M+H]+}。
在反应瓶中依次加入940.0g(86.6mmol),甲醇254mL,钯碳4.0g及甲酸铵16.3g(259mol),搅拌下回流反应2h(TLC跟踪)。用硅藻土过滤,滤饼用甲醇多次冲洗,合并滤液和洗液,浓缩得白色固体1021.1g,收率91%;1H NMRδ:7.55(s,1H),7.29(s,1H),6.79(s,1H),5.58(s,H),5.36(s,1H),5.15(s,1H),4.04(s,1H),3.73~3.81(m,3H),3.61(s,1H),2.06(s,3H);ESI-MSm/z: 270.4{[M+H]+}。
(8)(1R,3R,4R,7S)-3-(5-甲基胞嘧啶-1-基)-1-(4,4′-二甲氧基三苯代甲基氧代甲基)-7-羟基-2,5-二氧杂二环-[2.2.1]庚烷(11)的合成
在反应瓶中加入1024.0g(89.6mmol),氮气保护下加入无水吡啶180mL,4,4′二甲氧基三苯基氯甲烷(DMTCl)30.2g(89.5mol),搅拌下于室温反应4h(TLC跟踪)。加水(200mL)稀释,用乙酸乙酯(100mL)萃取,有机相用水(3×200mL)洗涤,浓缩后经硅胶柱层析(洗脱剂:A=2∶8)纯化得白色固体1138g,收率75%;1H NMRδ:6.88~7.49(m,14H),6.88(s,1H),5.67(d,J=3.0Hz,1H),5.39(s,1H),3.97~4.11(m,2H),3.28~3.40(m,10H),1.96(s,3H);ESI-MSm/z: 572.6{[M+H]+}。
(9)(1R,3R,4R,7S)-7-(三甲基甲硅烷基氧基)-3-(5-甲基-4-N-苯甲酰基胞嘧啶-1-基)-1-(4,4′-二甲氧基三苯代甲基氧代甲基)-2,5-二氧杂二环-[2.2.1]庚烷(12)的合成
在反应瓶中加入1110.0g(17.5mmol),氮气保护下加入无水吡啶87.5mL,三乙胺24.5mL和三甲基氯硅烷(TMSCl)3.5mL ,搅拌下于室温反应1h(TLC跟踪);加入苯甲酰氯2.2mL,于室温反应1h(TLC跟踪)。倒入200mL冰水中,用乙酸乙酯(2×50mL)萃取,合并萃取液,依次用水和饱和氯化钠溶液洗涤,无水硫酸钠干燥,浓缩得淡黄色固体12粗品14.0g,收率107%,直接用于下步反应;ESI-MSm/z: 748.8{[M+H]+}。
(10)(1R,3R,4R,7S)-3-(5-甲基-4-N-苯甲酰基胞嘧啶-1-基)-1-(4,4′-二甲氧基三苯代甲基氧代甲基)-7-羟基-2,5-二氧杂二环-[2.2.1]庚烷(13)的合成
在反应瓶中依次加入1213.0g(17.4mmol),THF 87.0mL,1mol·L-1四丁基氟化铵(TBAF)的THF(6.0mL)溶液,搅拌下于室温反应0.5h(TLC跟踪)。加水(100mL)稀释,用二氯甲烷(50mL)萃取,有机相依次用碳酸氢钠溶液和饱和氯化钠溶液洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析(洗脱剂:A=4∶6)纯化得白色固体1310.3g,收率88%;1H NMRδ:13.42(s,1H),8.29(d,J=6.0Hz,2H),7.82(s,1H),7.26~7.53(m,12H),6.88(m,4H),5.71(s,1H),4.47(s,1H),4.27(s,1H),4.00~4.10(m,2H),3.80~3.83(m,2H),3.80(s,6H),3.59(d,J=11.4Hz,1H),3.46(d,J=14.1Hz,1H),1.89(s,3H);ESI-MSm/z: 676.7{[M+H]+}。
(11)1的合成[7]
在反应瓶中依次加入1310.0g(14.8mmol),二氯甲烷74mL,氮气保护下加入N,N-二异丙基乙胺1.91g(14.8mmol),缓慢滴加入(二异丙基氨基)(2-氰基乙氧基)氯化磷4.7mL,搅拌下于室温反应8h(TLC跟踪)。加水(50mL)稀释,用二氯甲烷(2×50mL)萃取,合并萃取液,依次用碳酸氢钠溶液和饱和氯化钠溶液洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析(洗脱剂:A=3∶7)纯化得白色粉末110.5g,收率81%;1H NMRδ:12.38(s,1H),8.16(s,2H),7.86(s,1H),7.27~7.58(m,12H),6.89~6.94(m,4H),5.62(s,1H),4.49(s,1H),4.43(d,J=8.7Hz,1H),4.01(d,J=7.2Hz,1H),3.81(d,J=8.1Hz,1H),3.74(s,6H),3.44(s,5H),2.71(d,J=10Hz,1H),2.60(d,J=8.0Hz,1H),1.98(s,3H),1.19~1.3(m,11H);ESI-MSm/z: 876.8{[M+H]+}。
2 结果与讨论
在合成6和8时,需严格控制氢氧化钠水解的温度和时间,温度过高或水解时间过长,均会引起胸腺嘧啶核糖副产物的生成。
在11至13的合成过程中,文献方法采用将11先用TMSCl保护,再与苯甲酰氯进行反应,最后用氨水脱去三甲基硅烷,三步总收率38%。在重复该方法的实验中,我们发现氨水在脱去三甲基硅烷的同时水解掉了苯甲酰基,导致收率降低。为此本文将11先用TMSCl进行保护,再与苯甲酰氯反应得12,最后用TBAF选择性脱去三甲基硅烷得合成13,三步总收率由38%提高至88%。
本文设计了一条新的合成5-甲基胞嘧啶磷酸核糖的路线。与原工艺相比,缩短了合成路线,总收率从7.50%提高至21.0%。
[1] 李茂生,徐祥,梁华平,等.锁核酸研究进展[J].生理科学进展,2003,34(4):319-323.
[2] 余文辉,周小梅,周大桥,等.锁核酸捕获TaqMan探针实时PCR和PCR-RFLP检测乙型肝炎病毒基因变异的研究[J].中华微生物学和免疫学杂志,2007,27(11):977-982.
[3] Mads D S,Lisbet K,Torsten B,etal.α-L-ribo-configured locked nucleic acid (α-L-LNA):Synthesis and properties[J].J Am Chem Soc,2002,124(10):2164-2176.
[4] Koshkin A A,Singh S K,Nielsen P,etal.LNA (Locked Nucleic Acids):Synthesis of the adenine,cytosine,guanine,5-methylcytosine,thymine and uracil Bicyclonucleoside monomers,oligomerisation,and unprecedented nucleic acid recognition[J].Tetrahedron,1998,54(14):3607-3630.
[5] Horton D,Tindall C G.Methylene-insertion reactions with unsaturated sugars.Synthesis of 4-C-cycloproply-D-ribo-tetrofuranose derivatives[J].Carbohyd Res,1970,15(8):215-232.
[6] Kochkine A,Fensholdt J,Pfundheller H M.Improved synthesis of [2.2.1]bicyclo nucleosides[P].WO 0056746,2000.
[7] Michael M,Flemming G H,Jesper Wengel.3′-C-branched LNA-type nucleosides locked in anN-type furanose ring conformation:Synthesis,incorporation into oligodeoxynucleotides,and hybridization studies[J].J Org Chem,2004,69(19):6310-6322.
ANewSyntheticMethodof5-MethylcytosineLockedNucleicAcid
GU Wen, CHEN Bei, GE Min
(State Key Laboratory of Materials-Oriented Chemical Engineering,College of Biotechnology and Pharmaceutical Engineering,Nanjing University of Technology,Nanjing 210009,China)
A new synthetic route of 5-methylcytosine LNA{(1R,3R,4R,7S)-7-[2-cyanoethoxy(diisopropylamino)phosphinoxy]-l-(4,4′-dimethoxytrityloxymethyi)-3-(5-methyl-4-N-benzoylcytosine-l-yl)-2,5-dioxabicyclo[2.2.1]heptan (1)} was developed.1with the total yield of 21.0% was synthesized by a twelve-step reaction of substitution reaction and hydrolysis reaction,etc,using furanose 3-O-benzyl-4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribofuranose as the starting material.The structure was confirmed by1H NMR and MS.
LNA;ribofuranose;cytosine;synthesis
2013-06-20;
2014-06-06
国家自然科学基金资助项目(21072094/B0206)
顾问(1988-),男,汉族,江苏淮安人,硕士研究生,主要从事药物及药物中间体的合成研究。
葛敏,博士,教授,E-mail: minge88@gmail.com
O626.4;O629.1
A
1005-1511(2014)01-0668-04
