新型含七元醚环的脯氨酸衍生物的合成*
2014-08-30盛钊君杜志云董长治
盛钊君,杜志云,董长治,张 焜
(1.广东工业大学 轻工化工学院 天然产物及绿色化学实验室,广东 广州 510006;2.法国巴黎第七大学 ITODYS实验室,法国 巴黎 75205)
·研究论文·
新型含七元醚环的脯氨酸衍生物的合成*
盛钊君1,2,杜志云1,董长治1,2,张 焜1
(1.广东工业大学 轻工化工学院 天然产物及绿色化学实验室,广东 广州 510006;2.法国巴黎第七大学 ITODYS实验室,法国 巴黎 75205)
以L-谷氨酸和苯甲醛为起始原料,经7步反应合成了重要中间体(2S,5R)-1-苄基-5-(2-羟乙基)吡咯烷-2-羧酸叔丁酯(1),总产率19%;1经TBDMSCl保护羟基后,用10%Pd/C催化氢解去除苄基制得游离胺(2S,5R)-5-(2-二甲基叔丁基硅氧烷基乙基)吡咯烷-2-羧酸叔丁酯(3);3与Boc-β-Cl-ala,在碱性条件下偶合得氯代烃消去产物(2S,5R)-1-[(2-叔丁氧甲酰胺基)丙烯酰基]-5-(2-二甲基叔丁基硅氧烷基乙基)吡咯烷-2-羧酸叔丁酯(4),或在中性条件下偶合得(2S,5R)-1-[(R)-2-叔丁氧甲酰胺基-3-氯丙酰基]-5-(2-二甲基叔丁基硅氧烷基乙基)吡咯烷-2-羧酸叔丁酯(5);4或5经四丁基氟化铵去除TBDMS保护并发生关环反应合成了新型的七元醚环化合物(4R,7S,9R)-4-甲基-4-叔丁氧甲酰胺基-5-羰基-3-氧杂氮杂卓[1,4]并吡咯[1,2-d]-7-羧基叔丁酯(简称6);6于室温在氯仿中静置2d~3d自发转换为其构象异构体(4S,7S,9R)-6(简称6′,6/6′=1/9),其结构经1H NMR,13C NMR,2D NMR和HR-ESI-MS表征。
七元醚环;非肽药物;构象异构;合成
以生物活性肽为先导化合物,设计、合成非肽类小分子模拟物已成为药物化学领域的研究热点。相对肽类药物,非肽类药物能避免其在临床应用中的局限性,如细胞穿透能力差、体内稳定性差等,但同时,非肽类小分子药物又具有天然肽的特定构象以及生理活性,从而具有药用价值[1]。

SMAC/DIABLO(第二个线粒体来源的胱氨酸酶激活剂/低等电点IAP直接结合蛋白)是一种从线粒体中释放的凋亡抑制蛋白(IAPs)内源性拮抗剂[2]。研究发现,Smac通过N端Ala-Val-Pro-Ile四肽(AVPI,Ⅰ,Chart 1)与IAPs结合,从而抑制IAPs对细胞凋亡的抑制作用[3]。于是,以AVPI为先导化合物,设计和合成小分子Smac模拟物,并用于抗肿瘤药物成为近年的研究热点[4]。迄今为止,文献已报道多种IAPs拮抗剂[5],几种具有代表性的IAPs拮抗剂的骨架结构Ⅱ~Ⅳ见Chart 1,它们都是脯氨酸类似物,双环结构中的另一个环可以是七元[6-7]或八元[8]碳环,也可以是八元氮杂环[1,9]或七元氧杂环[10]。从这些结构出发,C端和N端连接不同的取代基团,可合成多种IAPs拮抗剂,并具有良好的活性。
为了丰富IAPs拮抗剂的种类,并发展出活性更佳的拮抗剂,本文报道了一种全新的含七元氧杂环的双环母核结构的合成方法。以L-谷氨酸和苯甲醛为起始原料,按文献[11]方法,经7步反应合成了重要中间体(2S,5R)-1-苄基-5-(2-羟乙基)吡咯烷-2-羧酸叔丁酯(1),总产率19%;1经叔丁基二甲基氯硅烷(TBDMSCl)保护羟基后,用10%Pd/C催化氢解去除苄基得游离胺(2S,5R)-5-(2-二甲基叔丁基硅氧烷基乙基)吡咯烷-2-羧酸叔丁酯(3);3与Boc-β-Cl-ala,在碱性条件下偶合得氯代烃消去产物(2S,5R)-1-[(2-叔丁氧甲酰胺基)丙烯酰基]-5-(2-二甲基叔丁基硅氧烷基乙基)吡咯烷-2-羧酸叔丁酯(4),或在中性条件下偶合得(2S,5R)-1-[(R)-2-叔丁氧甲酰胺基-3-氯丙酰基]-5-(2-二甲基叔丁基硅氧烷基乙基)吡咯烷-2-羧酸叔丁酯(5);4或5经四丁基氟化铵(TBAF)去除TBDMS保护并发生关环反应合成了新型的七元醚环化合物(4R,7S,9R)-4-甲基-4-叔丁氧甲酰胺基-5-羰基-3-氧杂氮杂卓[1,4]并吡咯[1,2-d]-7-羧基叔丁酯[(4R,7S,9R)-6,简称6];6于室温在氯仿中静置2d~3d自发转换为其构象异构体[(4S,7S,9R)-6,简称6′,6/6′=1/9](Scheme 1),其结构经1H NMR,13C NMR,2D NMR和HR-ESI-MS表征。
1 实验部分
1.1 仪器与试剂
Bruker AVANCEⅢ400NMR型核磁共振仪(CDCl3为溶剂,TMS为内标);LCT Premier XE型飞行时间质谱仪。
所用试剂均为分析纯,其中二氯甲烷和四氢呋喃用前经除水处理;柱层析硅胶(230目~400目)和薄层层析硅胶板(F254),Merck。
1.2 合成
(1)2的合成
在反应瓶中依次加入1500mg(1.64mmol)和二氯甲烷10mL,搅拌使其溶解;加入TBDMSCl 370mg(2.46mmol)和咪唑179mg(2.62mmol),用N,N-二异丙基乙胺(DIEA)调至pH9~10,于室温反应2h。用水洗涤,水相用乙醚萃取,合并有机相,依次用3%NaHSO4溶液、饱和NaHCO3溶液和饱和食盐水洗涤,MgSO4干燥,旋干溶剂后经硅胶柱层析[洗脱剂:A=V(环己烷)∶V(乙酸乙酯)=12∶1]纯化得无色油状液体2615mg,产率89%;1H NMRδ: 7.30~7.16(m,5H),3.89(d,J=14.10Hz,1H),3.74~3.57(m,3H),3.17~3.13(m,1H),2.86~2.79(m,1H),1.94~1.78(m,4H),1.63~1.47(m,2H),1.31(s,9H),0.86(s,9H),0.00(s,6H);13C NMRδ: 174.33,139.02,129.78,128.36,127.17,80.32,66.72,62.08,61.27,57.51,38.32,30.61,28.74,28.29,26.31,18.63,-4.95,-5.00。
(2)3的合成
在反应瓶中加入2330mg(0.79mmol)和乙酸乙酯10mL,搅拌使其溶解;加入10%Pd/C 66mg,通氢气反应过夜。用硅藻土过滤,滤液减压蒸除溶剂得无色油状液体3236mg,产率91%,直接进行下步反应;1H NMRδ: 3.72(t,J=6.48Hz,2H),3.63(dd,J=5.50Hz,8.88Hz,1H),3.17~3.10(m,1H),2.11~2.01(m,2H),1.92~1.76(m,3H),1.73~1.65(m,1H),1.46(s,9H),1.33~1.23(m,1H),0.89(s,9H),0.05(s,6H);13C NMRδ: 174.75,81.32,61.85,60.95,57.93,39.06,32.15,30.77,28.38,26.29,18.62,-5.00,-5.01。
(3)4的合成
在反应瓶中加入3330mg(1mmol)和二氯甲烷10mL,搅拌使其溶解;于0℃依次加入Boc-β-Cl-ala 240mg(1.07mmol),卡特缩合剂(BOP)486mg(1.10mmol)和DIEA 0.60mL,反应2.5h。依次用1%NaHSO4溶液、饱和碳酸氢钠溶液、水和饱和食盐水洗涤,无水MgSO4干燥,减压蒸除溶剂后经硅胶柱层析(洗脱剂:A=6∶1)纯化得无色油状液体4340mg,产率64%;1H NMRδ: 6.69(br,1H),5.77(br,1H),4.83(br,1H),4.51(br,1H),4.24(br,1H),3.80~3.54(m,2H),2.37~1.64(m,6H),1.46(s,9H),1.44(s,9H),0.88(s,9H),0.04(d,J=3.60Hz,6H);13C NMRδ: 171.99,152.96,136.55,100.08,82.18,80.78,61.28,60.94,58.11,37.35,30.15,28.57,28.25,27.25,26.26,18.57,-5.03,-5.10;HR-ESI-MSm/z: Calcd for C25H47N2O6Si{[M+H]+}499.3203,found 499.3180。
(4)5的合成
于0℃在反应瓶中依次加入二氯甲烷5mL,Boc-β-Cl-ala 82mg(0.36mmol)和缩合剂碳二亚胺盐酸盐(EDCI)116mg(0.61mmol),搅拌使其溶解;于0℃反应1h。加入3100mg(0.30mmol),于室温反应6h。依次用1%KHSO4溶液、饱和 NaHCO3溶液和饱和食盐水洗涤,无水MgSO4干燥,旋蒸脱溶后经硅胶柱层析(洗脱剂:A=7∶1)纯化得无色油状液体587mg,产率54%;1H NMRδ: 5.34(d,J=7.80Hz,0.39H),5.11(d,J=7.90Hz,0.40H),4.78~4.70(m,0.41H),4.66(t,J=6.30Hz,0.46H),4.57~4.51(m,0.47H),4.47~4.38(m,0.44H),4.32~4.28(m,0.48H),4.23~4.17(m,0.49H),3.80~3.54(m,4H),2.30~1.49(m,6H),1.45~1.40(m,18H),0.89~0.87(m,9H),0.07~0.03(m,6H);13C NMRδ: 171.32,171.12,168.69,155.15,154.71,82.96,81.62,80.51,80.39,61.41,60.94,60.87,60.63,57.93,57.15,53.20,44.24,38.18,36.86,30.47,29.96,28.58,28.25,28.17,27.36,26.27,18.60,18.57,-5.02,-5.06,-5.10;HR-ESI-MSm/z: Calcd for C25H48N2O6SiCl{[M+H]+}535.2970,found 535.2989。
(5)6的合成
在反应瓶中依次加入523mg(0.04mmol)和THF 5mL,搅拌使其溶解;加入TBAF0.047mmol的THF(1mol·L-1,47μL)溶液,于室温反应4h。旋蒸脱溶后经硅胶柱层析(洗脱剂:A=5∶1)纯化得无色油状液体610mg。
将6的二氯甲烷或氯仿溶液于室温静置约2d~3d得6′。
6:产率61%;1H NMRδ: 5.34(br,1H),4.51~4.40(m,2H),3.89(t,J=6.9Hz,2H),2.19~1.76(m,6H),1.68(s,3H),1.45(s,9H),1.40(s,9H);13C NMRδ: 171.83,170.70,153.90,87.54,81.40,80.29,62.80,61.78,56.25,35.52,32.05,28.62,28.30,27.68,23.67;ESI-MSm/z: 383.2{[M-H]-}。
6′:1H NMRδ: 6.00(br,1H),4.40~4.33(m,2H),3.92~3.80(m,2H),2.25~1.81(m,6H),1.78(s,3H),1.47(s,9H),1.44(s,9H);13C NMRδ: 171.34,170.55,154.06,89.54,81.91,79.73,62.72,61.40,55.56,35.05,31.26,28.67,28.31,27.86,23.84;HR-ESI-MSm/z: Calcd for C19H33N2O6{[M+H]+}385.2339,found 385.2345。
2 结果与讨论
2.14和5的合成
4和5的合成结果表明,Boc-β-Cl-Ala极易发生消除反应,即使在弱碱条件下,也容易生成消除产物,因此,5无法通过经典的偶合反应条件制得。本文通过改变实验条件,在无碱条件下最终合成了5。值得一提的是,单一化合物5的1H NMR和13C NMR谱图中却有两组相似信号,最初推测是反应过程中发生了消旋,后经TLC检测及HPLC分析等多重途径验证,此处无消旋发生。推测可能原因是由于5的分子内氢键导致特殊的环状结构形成,造成了同一位置的H或C的空间构象存在着两种可能,不同构象在NMR谱中化学位移不同,从而产生两组信号峰,而这种现象在之后的关环时消失。
2.2 七元醚环的形成
不管是4还是5在进行TBDMS脱保护时,关环反应同时发生,生成相同产物6。推测形成6的可能机理如Scheme 2所示。首先,5在TBAF的作用下脱去TBDMS基团,生成游离的O-,产生的氧负离子提供碱性条件,导致5发生消除反应生成烯胺中间体(A),接着A发生结构互变,生成对应的亚胺中间体(C)。然后,同一分子的羟基作为亲核试剂,进攻C=N的碳原子,通过亲核加成反应,最终形成七元醚环6。
2.3 核磁跟踪6转换为6′
在6的合成中,通过1H NMR跟踪(图1)发现,反应最初只生成单一构象6,若将6的氯仿溶液于室温静置2d~3d,6自发地发生构象转换,缓慢地变为其构象异构体6′,最终以10%的6和90%的6′的溶液稳定地存在。若将6溶于乙酸乙酯中,则不会发生该变化。因此,推测可能的原因是反应开始生成动力学稳定、但热力学不稳定的R构象产物,氯仿提供弱酸性环境引发了其反应,转变为热力学稳定产物6′。
高分辨质谱结果进一步确认了这一变化,6和6′的分子量相同,结合核磁数据,可以得知6和6′是一组构象异构体。
2.4 6′的二维核磁表征
通过多键碳氢关系(HMBC)核磁谱图(图2)也可进一步证明该七元醚环的形成。由图2可清晰观察到O的一侧亚甲基上Hc,与另一侧的季碳C1有偶合信号,这说明了该七元醚环的形成。
而6′的构象则可以通过二维核磁技术NOESY(图3)来确定。由于1为已知化合物,构象已经确定,分子中两个手性C所连接的Ha和Hb处于环的一侧。由图3可以清晰地观察到,七元醚环上的甲基H与另外两个手性C上的H有关联,这说明该甲基与其空间距离较近,处于环的同一侧,由此确定了6′的构象。
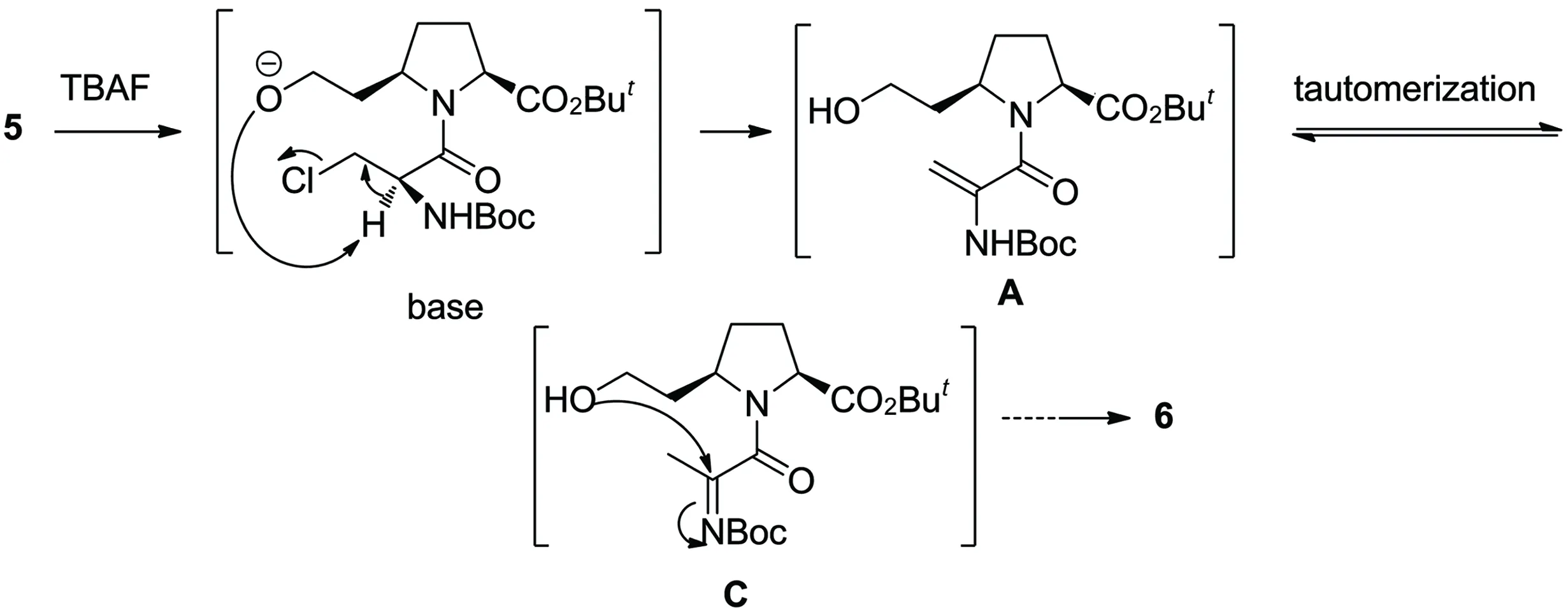
Scheme 2
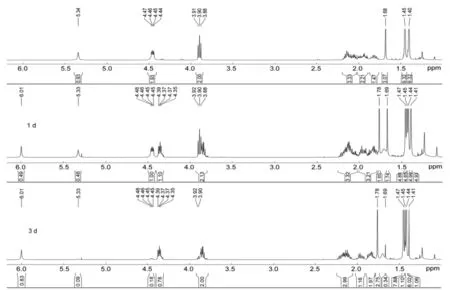
图1 6转化为6′的1H NMR谱图追踪Figure1 Conversion of 6 into 6′ tracing by 1H NMR

图2 6′的HMBC谱图(纯度90%,含10%6)Figure2 HMBC spectrum of 6′(purity 90%,containing 10%6)
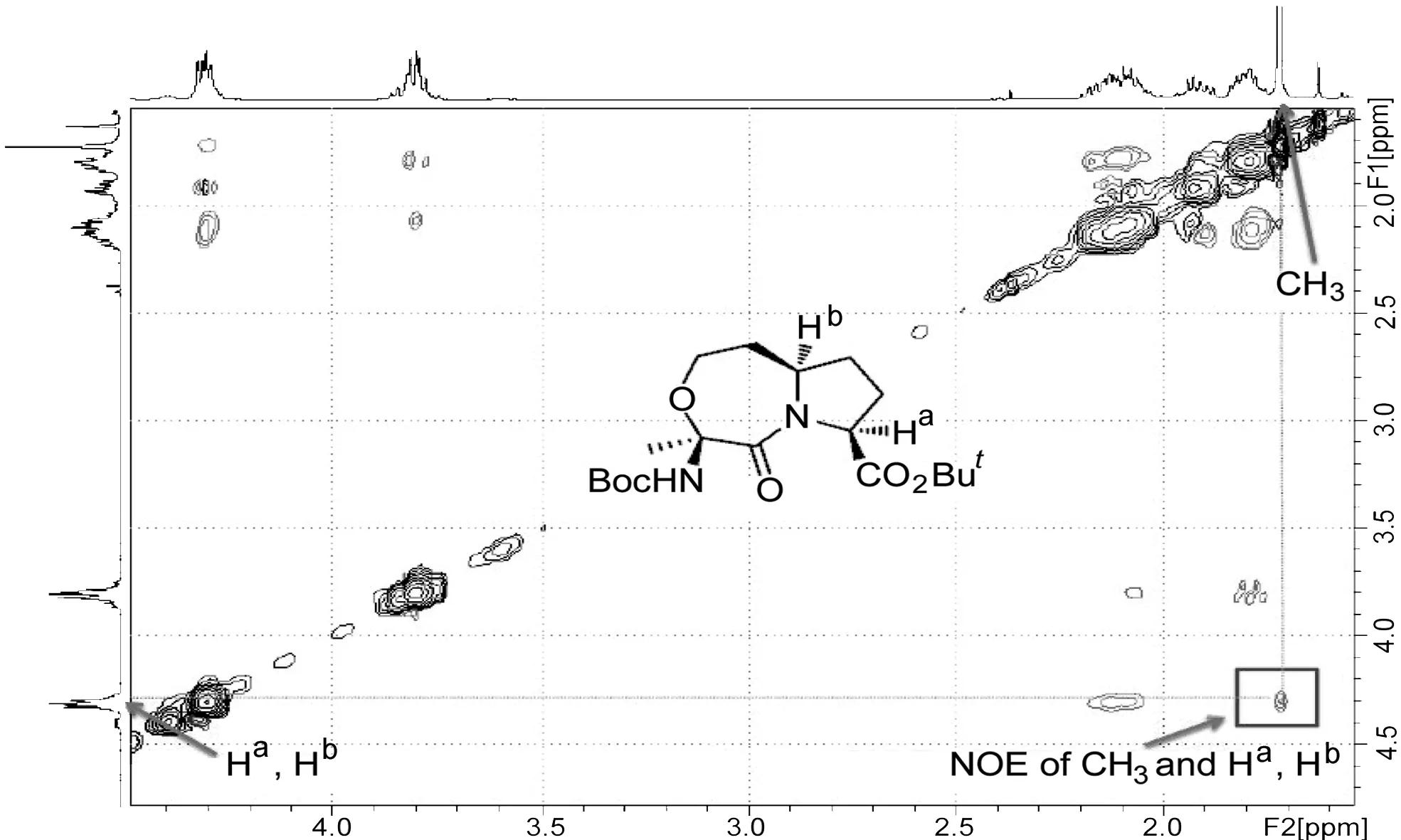
图3 6′的NOESY谱图(纯度90%,含10%6)Figure3 NOESY spectrum of 6′(purity 90%,containing 10%6)
3 结论
报道了一种全新的含七元醚环的脯氨酸衍生物的合成方法。同时,通过核磁技术观察到该化合物的两种构象异构体之间发生的自发转化,运用一维和二维NMR、HR-MS手段对其结构进行了表征。该全新结构可应用于IAPs拮抗剂的设计和合成中,通过变化其N-端和C-端的支链结构,有望得到全新系列的具有良好活性的IAPs拮抗剂。
[1] Y Peng,H Sun,S Wang.Design and synthesis of a 1,5-diazabicyclo[6,3,0]dodecane amino acid derivative as a novel dipeptide reverse-turn mimetic[J].Tetrahedron Letters,2006,47(27):4769-4770.
[2] G Wu,J Chai,T L Suber,etal.Structural basis of iap recognition by smac/diablo[J].Nature,2000,408(6815):1008-1012.
[3] E N Shiozaki,Y Shi.Caspases,iaps and smac/diablo:Mechanisms from structural biology[J].Trends in Biochemical Sciences,2004,29(9):486-494.
[4] H Sun,Z Nikolovska-Coleska,C Y Yang,etal.Design of small-molecule peptidic and nonpeptidic smac mimetics[J].Accounts of Chemical Research,2008,41(10):1264-1277.
[5] K Deshayes,J Murray,D Vucic.Protein-protein interactions,M Wendt.Vol.8[M].Berlin:Springer Berlin Heidelberg,2012.
[6] H Sun,Z Nikolovska-Coleska,C Y Yang,etal.Structure-based design of potent,conformationally constrained smac mimetics[J].Journal of the American Chemical Society,2004,126(51):16686-16687.
[7] H Sun,Z Nikolovska-Coleska,J Lu,etal.Design,synthesis,and evaluation of a potent,cell-permeable,conformationally constrained second mitochondria derived activator of caspase (smac)mimetic[J].Journal of Medicinal Chemistry,2006,49(26):7916-7920.
[8] H Sun,Z Nikolovska-Coleska,J Lu,etal.Design,synthesis,and characterization of a potent,nonpeptide,cell-permeable,bivalent smac mimetic that concurrently targets both the bir2and bir3domains in xiap[J].Journal of the American Chemical Society,2007,129(49):15279-15294.
[9] Y Peng,H Sun,Z Nikolovska-Coleska,etal.Potent,orally bioavailable diazabicyclic small-molecule mimetics of second mitochondria-derived activator of caspases[J].Journal of Medicinal Chemistry,2008,51(24):8158-8162.
[10] M Vamos,K Welsh,D Finlay,etal.Expedient synthesis of highly potent antagonists of inhibitor of apoptosis proteins (iaps)with unique selectivity for ml-iap[J].ACS Chemical Biology,2013,8(4):725-732.
[11] J S Petersen,G Fels,H Rapoport.Chirospecific syntheses of (+)- and (-)-anatoxin a[J].Journal of the American Chemical Society,1984,106(16):4539-4547.
SynthesisofNovelProlineDerivativesContaining7-MemberedEtherRing
SHENG Zhao-jun1,2, DU Zhi-yun1, DONG Chang-zhi1,2, ZHANG Kun1
(1.Laboratory of Natural Medicine and Green Chemistry,School of Chemical Engineering and Light Industry,Guangdong University of Technology,Guangzhou 510006,China;2.ITODYS Laboratory,Université Paris Diderot,Paris 75205,France)
A key intermediate(1)with the total yield of 19% was prepared by a sven-step reaction using L-glutamic acid and benzaldehyde as the starting materials.3was prepared by the protection of the hydroxyl followed by the removal of the benzyl by hydrogenolysis with 10%Pd/C from1.4and5were obtained by the coupling reaction of3with Boc-β-Cl-ala under alkaline conditions or neutral conditions,respectively.A novel proline derivative(6)containing 7-membered ether ring was synthesized by the removal of TBDMS with tetrabutylammonium fluoride and then cyclization from 4or5.6spontaneously conversed fromRconformation toSconformation in chlorform to give (4S,7S,9R)-6(6′,6/6′=1/9).The structures were characterized by1H NMR,13C NMR,2D NMR and HR-ESI-MS.
7-membered ether ring;nonpeptidic drug;conformational isomerism;synthesis
2014-03-27
国家自然科学基金资助项目(21172046)
盛钊君(1987-),女,土家族,湖南常德人,博士研究生,主要从事药物合成的研究。E-mail: shengliu165@hotmail.com
张焜,教授,Tel.020-39322270,E-mail: kzhang@gdut.edu.cn
O626
A
1005-1511(2014)05-0577-05
