外来吸附原子对Al13团簇的结构影响
2014-08-25王春婷李宝兴
陆 霞,孙 颖,王春婷,李宝兴
(杭州师范大学理学院,浙江 杭州 310036)
0 引 言
团簇是原子或分子向大块宏观物质转变过程中出现的特殊物态形式.它们的物理和化学性质随原子数而变化,所出现的奇异特性又展示出很多潜在的应用前景.因此,一直以来,科学家们采用各种手段和方法,付出极大努力研究团簇的物性.
Aln团簇的电子结构相对简单,随着原子数的增加表现出过渡金属的特性[1].Rao等人曾对Aln(n=2-15)团簇的几何结构、离化能与结合能进行了详细的研究,发现部分团簇表现出很好的稳定性.特别是Al13团簇,其结构为正20面体结构.在中性Al13与I原子反应的整个过程中,Al13团簇都保持着正20面的结构完整性,具有比I原子更大的原子亲和力,被称为“超级原子”.由于Al13团簇有39个价电子,容易与其它原子反应而达到满电子壳层结构,从而增强它的稳定性[2].过去几年,人们已经对铝团簇的性质进行了较多的研究,如用其它原子代替Al13团簇中的一个铝原子后性质所发生的变化[3]等.
对于Al13团簇的吸附研究虽有一些报道,如I和O等原子的吸附,但到目前为止还很有限.本文采用基于第一性原理的阿姆斯丹特密度泛函程序对Al13团簇吸附H、Li、Na、K、B、Al、Ga、C、Si、Ge、N、P、As等原子后的结构变化等进行了系统的研究,得到不同的吸附原子对它的结构影响规律以及它们的离子性质等.
1 计算方法
阿姆斯特丹密度泛函程序(Amsterdam Density Functional,简称ADF)采用了基于密度泛函理论的第一性原理方法[4],是目前国际上公认的用于团簇等研究的一种先进商业计算程序.在计算过程中,我们考虑了广义梯度近似(GGA),其中采用了Becke-Perdew(B-P)交换关联泛函.
用该程序首先研究了Al13团簇的稳定结构,得到了它的基态结构.在此基础上按不同吸附位置放置H、Li、Na、K、B、Al、Ga、C、Si、Ge、N、P、As等原子形成初始几何构型,并用上述程序对该初始构型进行结构优化,最后得到了各种稳定的吸附结构Al13+X.我们用该方法已经研究了一些团簇的吸附或掺杂性质[5-6],发现了一些有趣的现象.
2 结果与讨论
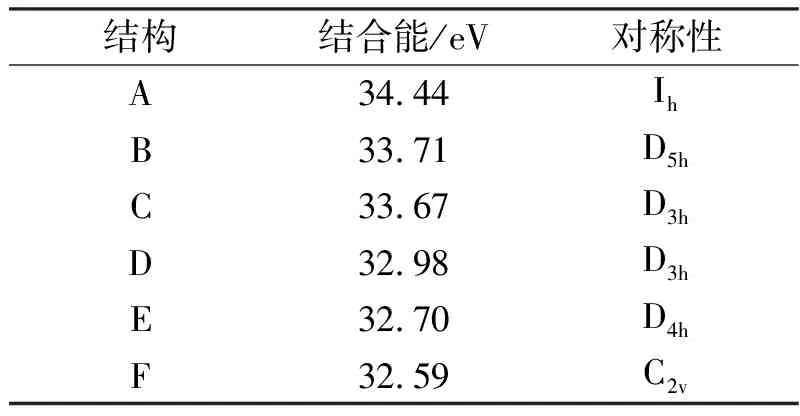
表1 Al13团簇6个稳定结构的结合能和对称性
我们采用了随机生成的方式得到了大量Aln团簇的初始结构.为了使各原子不会因为距离太近而重叠,也不会因为间距太远而造成收敛时间过慢,根据已经报道的Al团簇的原子间距范围,我们在自动生成的初始结构中将Al原子间的键长dAl-Al限定在2.1-3.5Å的范围内.同时,为了使初始结构具有多样性,我们把生成范围限定在长方体状、扁平体状和椭球形体状,原子可以随机分布在这三种体状的内部也可以分布在表面上.用ADF程序对这些自动生成的初始结构进行结构优化,发现许多结构不收敛,而又在很多情况下多个不同初始结构经过结构优化后变为同一个结构.比较结合能的大小,得到了如图1所示6个具有较大结合能的结构,它们的结合能和对称性如表1所示.最后我们对它们进行了频率计算,没有出现虚频,表明这些结构是稳定的.这6个结构的稳定性从A到F依次下降,其中A是Al13团簇的基态结构,具有Ih对称性,是典型的20面体结构.在目前所有的文献报道中,它是公认的基态结构.团簇的结合能包括静电能、动能、库仑能、交换关联能4部分.具体公式不再在这里一一列出.
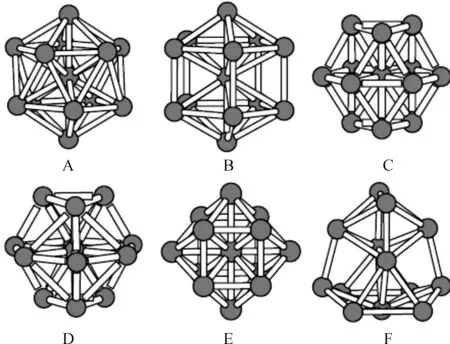
图1 Al13团簇的6个稳定结构

Al13团簇有Ih对称性,其表面有3个不同的位置:顶位、洞位和桥位.把H、Li、Na、K、B、Al、Ga、C、Si、Ge、N、P、As等原子分别放置这3个位置上产生初始结构,然后用ADF程序进行结构优化,最后得到它们的稳定吸附结构,它们的结合能见表2.
图2给出了氢和碱金属Li、Na、K的吸附结构.图中Al13+H(1)、Al13+H(2)和Al13+H(3)分别对应于氢在顶位、洞位和桥位的吸附结构.它们的结合能分别为3.59、3.88和3.90 eV,可见桥位为最佳吸附位置,但洞位几乎与之一样稳定.当Li原子吸附在Al13团簇上时,顶位结构有所改变,变为如图1中的Al13+Li(1)结构,但洞位和桥位结构类似于H原子的吸附结构(因类似于Al13+H(2)和Al13+H(3)结构,故没有画出图).当Na原子被吸附时,顶位结构又发生了改变,变成如图1中的Al13+Na(1)结构,同时洞位和桥位变成同一吸附结构,类似于Al13+H(2)结构(图没有画出).而K原子在顶位的吸附结构与Al13+H(1)结构类似,但不是很稳定,洞位和桥位结构优化后变为同一结构,并与Al13+H(2)结构相似.可见碱金属Li、Na、K吸附在Al13团簇上后,Li、Na原子在顶位的吸附结构与H原子的吸附结构各不相同,Li原子在桥位和洞位的吸附结构与H原子的吸附结构相似,但Na、K在洞位和桥位上的吸附结构经过结构优化后将变为同一结构,类似于结构Al13+H(2)结构.

表2 各原子吸附在Al13团簇上的结合能
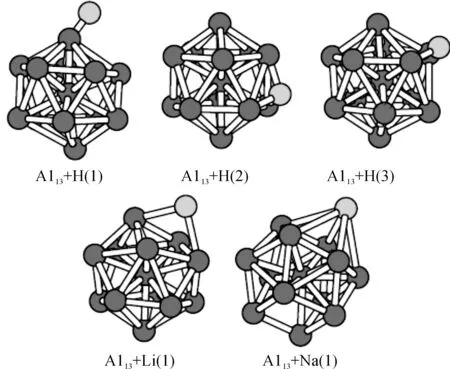
图2 Al13团簇吸附H、Li、Na、K原子结构图
图3给出了IIIA族元素B、Al、Ga的吸附结构.图3中的Al13+B(1)、Al13+B(2)和Al13+B(3)结构分别对应于B原子吸附在Al13团簇顶位、洞位和桥位的结构图.它们与H原子的吸附结构比较,有较大的差别,结构畸变非常明显,表明这些原子被吸附后将严重影响原Al13团簇的结构.洞位是B原子的最佳吸附位.Al原子吸附在Al13团簇上后实际成为Al14团簇结构.Al原子吸附在洞位和桥位上的优化结构是同一结构,而且是Al14团簇的基态结构,它比顶位的吸附结构要稳定0.16eV.Ga原子的吸附结构与Al的原子的吸附结构很相似,因此没有在图3中重复画出.
图4给出了IV族元素C、Si、Ge的吸附结构.图4中的Al13+C(1)、Al13+C(2)和Al13+C(3)结构分别对应于C原子吸附在Al13团簇顶位、洞位和桥位的结构图.它们与IIIA族元素的吸附结构比较,又有一些新的改变,表明不同族原子的吸附对原Al13团簇的结构影响是不同的.顶位是C原子的最佳吸附位.但当Si、Ge原子吸附在Al13团簇上后,洞位和桥位上的吸附经结构优化后变为同一结构,其结构类似于Al13+C(2)结构,比在顶位的吸附结构要稳定.对应Si和Ge,分别要稳定0.16 eV和0.13 eV.

图3 Al13团簇吸附B、Al、Ga原子结构图

图4 Al13团簇吸附C、Si、Ge原子结构图

图5 Al13团簇吸附N、P、As原子结构图
图5给出了V族元素N、P、As的吸附结构.图5中的Al13+N(1)、Al13+N(2)和Al13+N(3)结构分别对应于N原子吸附在Al13团簇顶位、洞位和桥位的结构图.很明显,原子吸附后,结构畸变比前面所有相应吸附结构还要明显.洞位是N原子的最佳吸附位.当P、As原子吸附在Al13团簇上后,顶位吸附结构与Al13+N(1)结构相似,而洞位和桥位吸附结构经结构优化后变为同一结构,如图5中的Al13+P(3)结构所示.
从以上的图示结果可以看出,不同原子吸附在Al13团簇上后,它们对Al13团簇的结构影响不同.具体可归纳为如下几点:1)氢原子对Al13团簇的影响最小,它几乎不改变Al13团簇的基本结构,但其它原子不同程度地使Al13团簇结构发生畸变.2)第二周期中的原子Li,B,C和N吸附在Al13团簇上的3个不同位置后,畸变程度不同,稳定性也不一样,并随着原子序号的增加而畸变程度增加;3)第三周期中的原子Na,Al,Si和P与第四周期中的原子K,Ga,Ge和As,如果它们属于同一族中,则它们对Al13团簇的结构影响相似,特别是它们吸附在洞位和桥位处时,经结构优化后都将变为同一吸附结构.
事实上,不同元素对Al13团簇的结构影响在定性上是可以理解的.如同一周期元素随着原子序号的增加对其结构改变越来越严重,这是因为同一周期中,从左往右,原子失去电子的能力减弱,获取电子的能力增强,结果是非金属性不断增强.Al13是金属团簇,吸附一个Al原子后成为Al14团簇,它的基态结构如图3中的Al13+Al(2).Al13团簇吸附Al原子后,原Al13团簇结构基本没有改变.另一方面,我们知道,Al13团簇有39个价电子,根据凝胶模型,它容易从其它金属原子中获取一个电子来达到满电子壳层结构,增强其结构的稳定性.特别是氢原子被吸附后,它提供一个外来电子,使该吸附结构达到满电子壳层结构,增强了稳定性.另外,金属原子被吸附后也可提供额外的电子,它们对Al13团簇的结构影响比较小.相反,由于非金属原子被吸附后获取电子的能力较强,所以它们会明显导致Al13团簇的结构畸变.也正因为如此,第三和第四周期中同一族中的元素,它们对Al13团簇的结构影响很相似.Mulliken布居分析表明纯Al13团簇中的中心原子从周围12个原子上各获得大约-1.0e的电荷,所以它带有大约-12e的电荷.当以上这些外来原子被它吸附后,大部分Al13+X吸附结构中各铝原子的电荷分布没有太大的改变.但B、C和N原子被吸附后,在最稳定吸附结构中的中心原子所带的电荷分别从原有约-12e降到-5.8e、-8.7e和-2.1e.其次是P和As原子,它们使中心原子的电荷量也分别降到-9.7e和-9.8e.这些外来原子被吸附后都获得了电荷,使得原Al13结构中的所有原子的带电量减少,其中中心原子所带的负电量和外围原子所带的正电量都相应减少.这些获取电子能力较强的外来原子都是非金属元素,它们明显改变了原Al13团簇结构中的电荷分布,破坏了原正20面体的结构平衡,所以这些吸附结构都存在明显的结构畸变.另外,不同原子的成键性质在这些吸附结构中也有所体现.如碳原子C被吸附后,由于它的共价性很强,所以正如图4中的Al13+C(1)结构(3个吸附结构中最稳定的结构),它与Al13团簇中的4个Al原子形成明显的共价键.再如氮原子N被吸附后,它的最稳定结构为Al13+N(2),从图可以看出N与Al13团簇中的3个Al原子也形成较明显的共价键.

图6 Al13团簇和Al13+C团簇最高4个占据轨道的电子云图
图6是Al13团簇和它吸附碳原子C后的4个最高占据轨道对应的电子云图.紫色小球和灰色小球代表铝原子Al和碳原子C,呈不规则的蓝色和红色片状部分代表电子云.大写英文字母A对应的图为它的HOMO轨道图,字母B、C、D对应的轨道能级依次降低.吸附碳原子C后对应的4个轨道电子云图用小写英文字母a、b、c、d来表示.观察图6可知,Al13团簇的4个最高占据轨道,有两个简并.这4个轨道中绝大部分原子形成π键或大π键,而吸附碳原子C后的最大变化是它的成键特征,从图a、b和c容易发现,吸附碳原子C后,大部分键变为σ键.另一方面,比较这4对电子云图,相对变化较小的是D和d图,它们在各自4个能级中能量最低,可见吸附外来原子对其影响最大的是接近HOMO能级的几个轨道.因此,容易理解吸附碳原子后结构会更稳定一些.类似地,吸附氮原子N后,其结构也有明显的改变(见图5).这主要是因为吸附氮原子N后,Al13团簇中HOMO附近的几个轨道,成键性质发生了明显的变化,σ键的数目增加,稳定性增加.
3 小 结
我们用基于密度泛函理论的第一性原理计算程序,对Al13团簇的原子吸附特性(吸附H、Li、Na、K、B、Al、Ga、C、Si、Ge、N、P、As)进行了系统的研究,研究表明不同周期中的元素对结构的影响是不同的.第二周期中的原子对Al13团簇结构的影响随着原子序号的增加而逐步增加,第三和第四周期同一族中的原子的结构影响相似,特别是它们吸附在洞位和桥位处,经结构优化后变为同一结构.这些性质与Al13团簇本身和吸附原子的成键等性质有关.
[1]彭平,李贵发,郑采星,等.Aln(n=3,4,6,13,19)团簇的结构稳定性与形态演化[J].中国科学:E辑技术科学,2006,36(9):975-982.
[2]Rao B K, Jena P. Evolution of the electronic structure and properties of neutral and charged aluminum clusters: A comprehensive analysis[J]. J Chem Phys,1999,111(5):1890-1904.
[3]陈莉莉,卢其亮,李永.氧原子在X+Al12(X=C,Si,P+)团簇吸附性质的研究[J].原子与分子物理学报,2010,27(5):863-868.
[4]Lenthe E, Baerende E J, Optimized Slater-type basis sets for the elements 1-118 [J]. J Comput Chem,2003,24(9):1142-1156.
[5]顾娇娇,李宝兴,马志伟.氮掺杂C20富勒烯的几何结构和稳定性[J].杭州师范大学学报:自然科学版,2013,12(2):140-144.
[6]马志伟,李宝兴,顾娇娇.CmNn(m,n=1-10,4≤m+n≤11)团簇的结构和稳定性研究[J].杭州师范大学学报:自然科学版,2013,12(4):359-364.
