基于固体酸的纤维素非均相催化糖化的研究进展
2014-08-08张颖诗王艳万金泉马邕文连洁
张颖诗,王艳,万金泉,马邕文,连洁
(华南理工大学环境与能源学院,广东 广州 510006)
生物质既能够替代化石能源,又能够通过光合作用固定无机碳,实现二氧化碳双重减排,满足可持续发展的要求,被认为是具有广阔发展前景的替代能源[1]。在众多生物质资源中,木质纤维因具有来源广泛、不与食物供应形成竞争、避免引起粮食危机等优势而受到广泛关注。纤维素作为木质纤维的主要组成部分能够转化成葡萄糖、果糖、5-羟甲基糠醛、乙酰丙酸以及包括山梨醇、甘露醇、乙二醇在内的多元醇等多种重要能源物质和基础平台化合物。上述化合物的生成包括两大阶段:首先是纤维素水解生成葡萄糖,进而葡萄糖发生异构化、脱水、再水合、加氢、氢解等不同反应生成不同的产物。由此可见,葡萄糖是纤维素转化过程中的一种重要的中间产物,其他的产物都能够由它经过不同的反应制备而成,所以纤维素定向转化成葡萄糖是实现纤维素高效转化利用的关键。
传统的纤维素水解均相催化体系主要采用无机酸作为催化剂,葡萄糖产率高,但存在副产物种类多、腐蚀设备、产物与催化剂难以分离等问题。而使用生物酶水解纤维素尽管能耗较低、糖产率高、无污染,但是酶对反应环境敏感,容易失活,而且难以接触纤维素结晶结构导致反应时间长、效率低,以上不足限制了酶法水解纤维素的推广应用[2]。与均相催化体系相比,非均相催化剂易于分离、回收,能有效避免对设备的腐蚀,同时能够通过调整比表面积、孔径尺寸等物理结构和嫁接酸性中心、吸附中心等活性官能团以满足催化反应的需求而受到关注,目前非均相催化剂主要以磺化固体酸、沸石、金属氧化物、负载金属、杂多酸为主。本文以实现纤维素高效转化为葡萄糖为目的,从催化剂物理化学结构对催化活性的相互作用关系的角度出发,综述上述5种主要类型的非均相催化剂的研究进展、存在的弊端以及今后的发展方向。
1 磺化固体酸
磺化固体酸催化剂指含有磺酸基团、固体表面提供酸催化中心的催化剂,是催化纤维素水解的固体酸催化剂中研究最为广泛、深入的一类催化剂。表1列举出近年来的研究成果,根据被嫁接磺酸基团的材料,可分为炭基磺化固体酸和磺化聚合物固体酸两类。
1.1 炭基磺化固体酸
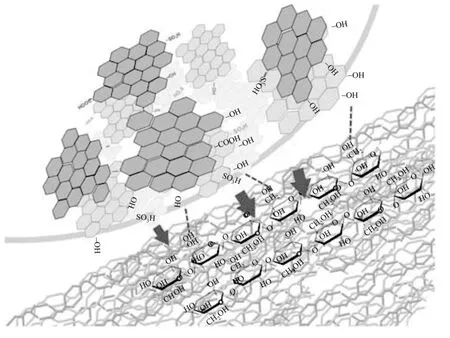
图1 炭基磺化固体酸催化水解纤维素机理[6]
炭基磺化固体酸是将一些有机材料经过碳化处理,同时使其含有磺酸基团的一类固体酸。炭基磺化固体酸的合成思路有两种:一种是部分碳化带有磺酸基团的多环芳香化合物;另一种是磺化经过碳化处理的有机化合物。Hara等[4]根据第一种合成思路,先磺化处理萘、蒽、二萘嵌苯等煤焦油、石油中大量存在的多环芳香化合物,然后在浓硫酸作用下加热进行不完全碳化。合成的催化剂是含—SO3H的多环芳香碳环以无序方式堆积而成的无定形碳,其最高—SO3H密度可达4.90mmol/g。但是由于连接在无序堆积碳环上的—SO3H大部分处在催化剂的内部,纤维素难以与之接触,所以有效—SO3H密度远低于所测密度。此外,—SO3H密度高且与芳香环上的碳紧密联系导致其他具有吸附性能和酸性的活性官能团难以通过连接芳香环的碳而嫁接到催化剂上,加大了嫁接活性官能团以提高催化剂活性的难度。因此,目前研究的炭基磺化固体酸催化剂大多按第二种合成思路合成,首先通过高温加热使带—OH有机化合物如葡萄糖、纤维素等糖类不完全碳化,再通过浓硫酸处理将—SO3H嫁接在碳环。这种方法合成的催化剂一般都含有—OH、—SO3H、—COOH三种活性官能团(图1),其中—OH作为吸附中心能与纤维素糖苷键的氧原子形成氢键,将其吸附在催化剂表面,而不能与葡萄糖形成氢键发生吸附,催化剂对纤维素和葡萄糖的选择性吸附避免了葡萄糖的进一步酸催化反应,有利于提高葡萄糖的选择性;—SO3H、—COOH作为酸中心(酸密度为0.5~2.39mmol/g)在水中解离出H+进攻糖苷键上的氧原子使其迅速质子化,纤维素糖苷键进而断裂使纤维素解聚。这3种活性官能团均具有亲水性,既有利于催化剂在水中的均匀分布,又有利于携带低聚糖的水进入催化剂内部,与其中的酸中心接触反应,从而提高了酸的利用率和纤维素的水解糖化效率[5]。不同的有机化合物合成的炭基磺化固体酸虽然化学结构相似,但是物理结构、活性官能团密度差异是对催化效果影响的主要因素。

表1 不同磺化固体酸催化剂催化纤维素
高比表面积是提高纤维素与催化剂的碰撞频率、促进纤维素水解的重要因素。磺化活性炭(sulfonated activated-carbon,AC-SO3H)是在比表面积(941m2/g)上有着明显优势的一种炭基磺化固体酸,与其他碳源为纤维素(sulfonated microcrystalline cellulose-based catalyst,MCC- SO3H)[7]、葡萄糖(sulfonated glucose-based catalyst,GC-SO3H)[8]、蔗糖(sulfonated sucrose-based catalyst,SC-SO3H)[9]、酚类残渣(sulfonated phenolic residue-derived catalyst,PRC-SO3H)[10]、玉米壳水解残渣(sulfonated corncob hydrolyzed residue- derived catalyst,CRC-SO3H)[11]的低比表面积(<2m2/g)炭基磺化固体酸相比,催化纤维素效果显著。但在活性炭制备过程中产生的灰分会覆盖多孔结构表面,降低催化剂比表面积。Pang等[12]通过使用低浓度硝酸溶解表面灰分来提高活性炭比表面积,结果催化活性明显提高。高比表面积往往由多孔结构导致,纤维素的尺寸一般为10~20nm,不同类型孔结构对纤维素进出孔的限制有所差异,所以对催化剂水解效果的影响也有所差异。Zhang等[13]以木质素合成出孔径为6~8nm的木质素磺化催化剂(lignosulfonate-based catalyst,LF),由于催化剂的孔径小于纤维素,纤维素难以自由出入孔结构与孔中活性中心接触,所以LF催化效果比不上孔径分布在1~10000nm范围的AC-SO3H。Wang等[12]合成的催化剂磺化有序介孔碳(sulfonated ordered mesoporous carbon,CMK-3-SO3H)同时具有高比表面积和有序介孔结构(2~50nm),纤维素能够进入介孔接触其中的活性中心,而产生的葡萄糖则容易从介孔结构离开,由此实现纤维素的选择性接触,葡萄糖产率高达74.5%。
活性官能团密度对催化剂活性的发挥有重要影响。Jiang等[8]合成出以葡萄糖为碳源的催化剂(glucose-based catalyst,GC-SO3H),研究发现葡萄糖碳化程度低时催化剂保留高密度—OH,表现出对纤维素强吸附力,使葡萄糖产率最高达74%。而Suganuma等[14]以聚氯乙烯(PVC)为碳源合成磺化催化剂(sulfonated PVC-based catalyst,PVC-SO3H)催化水解纤维二糖,由于催化剂不 含—OH,很难吸附纤维二糖,所以无法表现出高催化活性,葡萄糖产率仅为30.1%。高密度酸中心能提供足够的酸保证发生纤维素水解反应,而炭基磺化固体酸可通过提高酸中心—COOH和—SO3H的密度实现酸密度提高。Zhang等[13]用低浓度硝酸处理活性炭,既能氧化活性炭表面的内酯基、醚基、醌基等官能团为—COOH,又能溶解活性炭表面灰分使更多—SO3H嫁接到活性炭上,催化剂的酸密度从1.58mmol/g提升至2.23mmol/g,葡萄糖产率提升到62.2%。Suganuma等[14]合成的PVC-SO3H虽然不含有—OH,但碳环上空位的碳有利于嫁接更多的—SO3H,且磺化反应为亲电反应,PVC-SO3H结构中的碳以sp3杂化为主,平均电荷密度更大,更有利于亲电反应的发生,所以PVC—SO3H上的—SO3H密度达到2.3mmol/g,大于其他炭基磺化固体酸。虽然增加—COOH、—SO3H在炭基磺酸固体酸上的数量都能实现酸密度的提高,但是对于这两种活性基团在催化剂上的数量分布是否存在相互制约以及它们对催化效果的共同影响关系,还有待深入研究。
—SO3H释放出H+后形成的—SO3-在含水酸性介质中会发生水解而脱落,由此造成的—SO3H损失是炭基磺化固体酸的应用难点。在不同的反应介质中—SO3H的损失情况有所差异,Onda等[15]以水为反应介质,研究发现AC—SO3H经过一次反应,溶脱在水中的SO42-含量达1.1mmol/L,对催化剂进行水热处理后—SO3H的稳定性显著提高,仅有不足0.03mmol/L的SO42-在反应中溶脱[16]。Liu[9]和Qi[17]等为实现纤维素的溶解,以离子液体[BMIM]Cl作为反应介质,反应中[BMIM]Cl与—SO3H发生离子交换生成—SO3[BMIM],导致水解液中溶脱出18.9%SO42-,损失情况比在水中反应严重。于是,Fu等[18]通过将[BMIM]Cl嫁接在用竹粉合成的磺化固体酸上[ionic liquid (IL)-grafting biochar sulfonic acid catalyst,BC-SO3H-IL],在保留[BMIM]Cl纤维素溶解能力的同时使反应在水中进行,避免了—SO3H的严重损失。此外,[BMIM]Cl与—SO3H通过形成分子内部氢键可以固定—SO3H减少其 脱落。
炭基磺化固体酸经过简单过滤即可与水解液分离,但催化剂表面残留未反应的纤维素会对其后续催化效果产生影响,Fu[19]和He[20-21]等的研究组提出在催化剂体系中引入带磁性的Fe,分别合成出Fe3O4-SBA-SO3H、Fe/C-SO3H及Fe3O4/C-SO3H,通过磁场彻底将催化剂与表面残渣分离。
1.2 磺化聚合物固体酸
磺化聚合物固体酸是指通过磺化反应将—SO3H嫁接到具有苯环结构的聚合物上而形成的一类磺化聚合物催化剂,区别于炭基磺化固体酸,其合成过程不需要经过碳化处理,化学结构中不含有—OH,仅有个别催化剂含有—COOH。
磺化聚合物固体酸的比表面积一般较大,所以孔结构对水解效率的影响更大。孔径大于50nm的磺酸树脂[16](Amberlyst 15)可使纤维素与孔内部的酸性中心—SO3H接触发生水解。而Matsuda等[22]合成的微孔孔径小于2nm的磺化金属有机骨架材料(sulfonated material from institute Lavoisier,MIL-101-SO3H)仅使纤维素分子链的一端进入催化剂的孔中与其中的酸性中心接触,催化效果不理想。因此,具有大于纤维素直径的介孔结构或者大孔结构的聚合物被看作是适合用于合成磺化聚合物的 原料。
磺化聚合物固体酸一般含有—SO3H而不含—COOH,所以通过提高—SO3H密度及引入—COOH可实现酸密度提高。而酸强度受催化剂平均电荷密度影响,当含有吸电子基团时电荷密度降低,使H+解离程度提高,表现出更高酸强度。Liu等[23]磺化处理聚二乙烯基苯合成磺化聚二乙烯基苯(sulfonated polydivinylbenzene,PDVB-SO3H)并将其与超强酸HSO3CF3反应,引入强吸电子基团(PDVB-SO3H-SO2CF3),在PDVB-SO3H合成过程中,PDVB的C=C键被氧化成—COOH,催化剂由—COOH和—SO3H共同提供的酸密度达3.50mmol/g,而—SO2CF3的引入降低了催化剂的电荷密度,使催化剂的酸强度明显提升,与此同时,—SO2CF3占用了原本可以嫁接—SO3H的C,催化剂的酸密度降低到3.34mmol/g。该催化剂中提高酸密度、酸强度存在的竞争关系是制约其催化活性的重要因素,因此如何实现二者平衡、使催化剂发挥最大效能是值得研究的问题。
缺少吸附中心是磺化聚合物固体酸催化效果比炭基磺化固体酸差的主要原因,使聚合物具有强电负性基团并以此作为吸附中心是解决这一问题的方向。Pan等[24]合成含有强电负性的—Cl的磺化氯甲基聚苯乙烯树脂(sulfonated chloromethyl polystyrene resin,CP-SO3H),通过—Cl与纤维素糖苷键中的—OH形成氢键来吸附纤维素,虽然葡萄糖与纤维素均具有—OH,但纤维素中—OH个数多于葡萄糖,所以催化剂对纤维素的吸附能力强于葡萄糖,因此—Cl对纤维素具有选择性吸附能力,在提高纤维素水解效率的同时也提高了葡萄糖的选择性。研究发现,—Cl在数量较少时催化剂的选择性吸附表现明显,但当含大量—Cl时就有足够的吸附中心同时吸附纤维素和葡萄糖,所以催化剂中强电负性基团的种类、数量与纤维素选择性吸附间的关系还需深入研究。
磺化聚合物固体酸也存在—SO3H损失的难题。研究表明,Amberlyst 15热稳定性差,在200℃反应24h后水解液呈深褐色,SO42-溶脱的数量与AC-SO3H相当[16]。Essayem等[25]合成含亲水性硅氧结构的磺化有机硅催化剂(sulfonated ordered mesoporous organosilica,C-SBA-SO3H),研究表明,在水热条件下催化剂结构中Si-(CH2)3-SO3H和Si-(CH2)3-SH的C—Si键发生断裂,导致—SO3H的严重损失。Fan等[26]在离子液体[BMIM]Br中研究磺化聚苯乙烯-二乙烯苯[sulfonated poly(styrene-co- divinylbenzene),SPS-DVB-SO3H]的催化活性,发现[BMIM]Br与SPS-DVB-SO3H发生离子交换生成—SO3[BMIM],如图2,虽然—SO3[BMIM]在反应后仍然保留在催化剂中,但是H+的损失使催化剂的酸密度大幅下降,葡萄糖产率从37%下降至7%。所以反应后将催化剂浸泡于硫酸溶液进行再生,成为保持催化剂活性的必要手段。
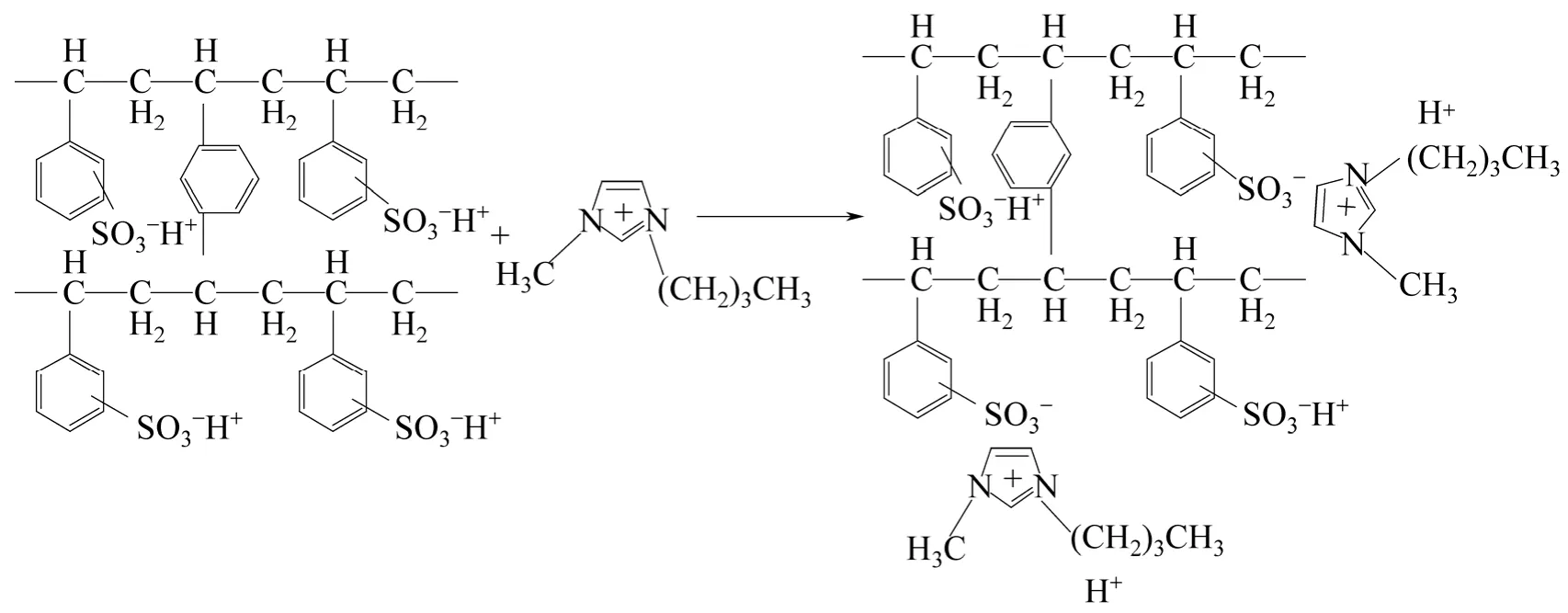
图2 SPS-DVB-SO3H和[BMIM]Br的离子交换反应[26]
综上所述,磺化固体酸催化剂中,高密度酸性中心(最高3.34mmol/g)能确保催化剂的酸催化活性,高密度吸附中心(—OH或—Cl)能够保证催化剂与纤维素充分接触和对纤维素的高选择性吸附,而高比表面积以及合适的孔径尺寸能够提高催化剂与纤维素的碰撞频率,但活性官能团间的制约关系是影响提升催化剂活性的关键,且酸中心(—SO3H)损失导致的催化活性降低是阻碍其应用的最大难题。
2 氢型沸石
沸石是硅铝组成的多孔矿物质,其多孔结构能容纳Na+、K+、Mg2+等阳离子,而容纳H+的沸石称为氢型沸石。氢型沸石除了本身具有来自末端硅羟基SiOH、桥式羟基Si-(OH)-Al、非骨架铝的铝羟基AlOH中酸性羟基的布朗斯特酸中心以及来自三配位铝原子的路易斯酸中心之外,还有与铝氧四面体中氧相连的H+,在纤维素的催化水解过程中能够为催化反应提供充足的酸催化活性(0.18~1.36mmol/g),同时氢型沸石105~429m2/g的高比表面积也确保其与纤维素的高碰撞频率。影响氢型沸石催化纤维素性能的主要因素是氢型沸石的硅铝摩尔比(Si/Al)。
氢型沸石化学结构Si/Al会直接影响催化剂的亲水性和酸密度。氢型沸石的亲水性源于化学结构中的—OH,而大多数—OH与Al相连,所以低Si/Al(<20)的氢型沸石相对铝含量较高,含有较多亲水性—OH,而高Si/Al(>20)的氢型沸石相对铝含量低,—OH数量少,表现出憎水性,有利于吸附纤维素,因此高Si/Al的氢型沸石表现出更高的催化效率。Onda等[15]的研究印证了这一结论,见表2。 他们对比不同Si/Al比氢型沸石的催化效果,结果表明H-ZSM5(Si/Al=75)和H-β(45)氢型沸石催化剂葡萄糖产率均高于低亲水性的H-β(12)和丝光沸石(10)。氢型沸石中的布朗斯特酸主要来源于桥式羟基Si-(OH)-Al、铝羟基AlOH,提高Si/Al、降低铝的相对含量,则降低催化剂的酸密度,在Onda的研究中,氢型沸石的酸密度随Si/Al的增高而降低。氢型沸石的催化效果受纤维素的吸附能力和酸密度影响,但是催化表现与憎水性的排序相一致,可见吸附能力对催化剂催化效果的影响比酸密度更大。
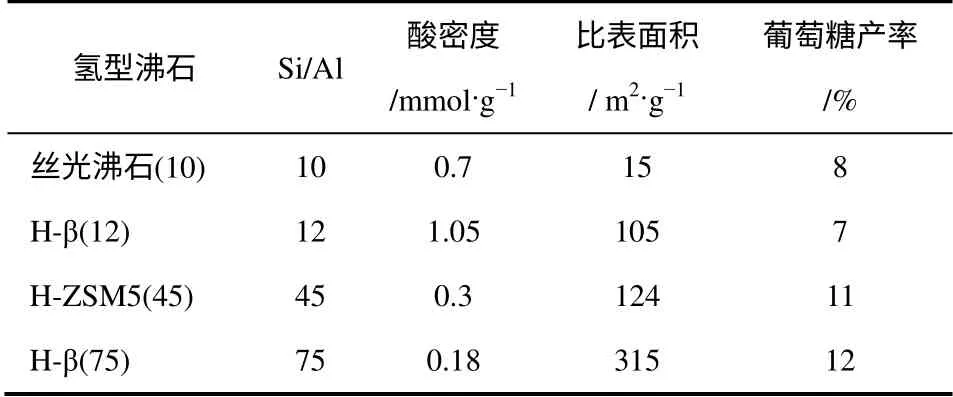
表2 氢型沸石特征参数及催化纤维素葡萄糖产率[15]
在保持氢型沸石Si/Al以保持憎水性的同时,可以通过嫁接酸性基团补偿酸密度的损失达到改善催化性能的目的。Yang等[27]采取氢型沸石H-USY与甲基巯丙基硅烷反应来嫁接巯基,然后用过氧化氢氧化巯基为—SO3H的方法,实现—SO3H嫁接,嫁接后催化剂的酸密度从0.26mmol/g上升至0.29mmol/g,使其水解纤维素的还原糖产率提高10%。
氢型沸石的微孔结构是催化剂内部活性中心作用发挥的制约因素。Yang等[28-29]通过草酸与H-USY发生脱铝作用产生羟基缺陷使催化剂的微孔扩大为介孔来促进纤维素进入催化剂内部,充分发挥其孔内酸性中心的催化活性,使H-USY催化半纤维素的还原糖产率从原来的5%提升至55%。若反应在离子液体中进行,氢型沸石孔中酸性羟基上的质子与[BMIM]+离子交换而释放出H+,扩大催化剂孔径则能使[BMIM]+更容易进入孔中,H+释放速率更高,催化纤维素水解活性更强。
虽然氢型沸石的憎水性、酸密度对其催化活性有一定的影响,但是孔径尺寸是氢型沸石充分发挥催化活性的最大障碍,在氢型沸石中引入介孔结构对其催化活性的提升效果高于引入酸性中心,因此有效改善氢型沸石的孔结构、提高纤维素在催化剂中的通过能力是保障其催化水解效果的重要方向。
3 金属氧化物
含有高价态过渡金属元素的金属氧化物有接受电子的能力,表现出路易斯酸性,能用于催化蔗糖、纤维二糖、纤维素的催化水解反应。然而路易斯酸性弱于布朗斯特酸性,所以路易斯酸用于催化纤维素水解的效果也普遍低于布朗斯特酸。金属氧化物只有路易斯酸中心,因此催化水解效果不理想。Domen等[30]采用Nb2O5·nH2O催化蔗糖和纤维二糖,由于酸密度仅为0.4mmol/g,获得的葡萄糖产率均为0。于是他们通过高温煅烧Li2CO3、Nb2O5和MoO3制备出过渡金属氧化物LiNbMoO6,然后用硝酸中的H+置换LiNbMoO6的金属离子Li+,在催化剂中引入布朗斯特酸制得HNbMoO6,酸密度提高到1.9mmol/g,催化淀粉和纤维素时分别获得20%和0.5%的葡萄糖产率。虽然HNbMoO6的酸强度有所提高,但是比表面积降低至5m2/g,而且层状结构的间距仅为0.553nm[31],远小于纤维素直径,纤维素难以进入催化剂的层状结构与其中的酸中心接触,抑制了催化活性的发挥。
尽管相关研究证实了金属氧化物对纤维素的催化水解能力,但是金属氧化物的弱酸性以及阻碍纤维素进入的层状结构决定其较弱的催化活性,所以金属氧化物在催化纤维素水解方面的研究和应用都较少。
4 负载金属
负载金属指将贵金属Ru、Pt负载于惰性载体用于催化纤维素水解加氢产多元醇的一类催化剂,而纤维素转化为多元醇的过程首先是使纤维素水解产生葡萄糖,所以Fukuoka等[32-33]推测负载金属催化剂在催化加氢反应的同时也起催化水解的作用,于是考察了负载钌的介孔碳材料(Ru/CMK)和负载铂的炭黑材料(Pt/BP2000)催化水解球磨后纤维素的催化活性,葡萄糖产率分别为34.2%和12.9%。Fukuoka等对Ru/CMK的催化机理有两种推测:一是Ru在催化体系中以RuO2·2H2O的形态出现,其中的结合水解离产生H+表现出布朗斯特酸性;二是Ru以三价态存在具有吸收电子能力,提供路易斯 酸性。
负载金属催化纤维素水解在190~230℃的高温下进行,反应过程能耗大,同时负载金属催化剂为弱酸性,用红外光谱检测催化剂对吡啶吸收峰值来表征酸密度,没有出现吸收峰,因此水解时间较长,导致葡萄糖水解副反应的发生,使水解产物分布广、葡萄糖产率偏低,所以在催化纤维素糖化方面的研究较少,而多用于氢气中催化纤维素水解加氢的连续反应。
5 杂多酸
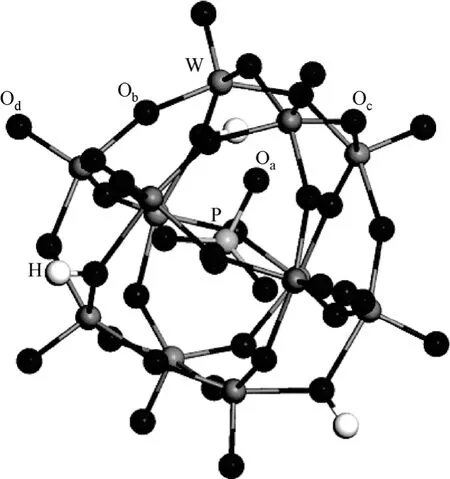
图3 H3PW12O40 结构图[36]
Keggin结构杂多酸是杂原子呈四面体配位、配原子呈八面体配位的一类含氧多酸,其杂多阴离子的独特“笼型”结构(图3)导致表面电荷密度低、 对抗衡阳离子H+的引力小,于是H+的活动性大,在有机溶剂或水中容易解离,表现出强布朗斯特酸酸性。Shimizu等[34]的研究表明杂多酸H3PW12O40、H4SiW12O40的酸强度超过硫酸和高氯酸。由于H3PW12O40具有强酸性而且完全溶于水,均相催化纤维素水解,在150℃下反应2h即能获得18%的还原糖产率,而硫酸仅能获得10%。由于是均相催化,所以H3PW12O40反应后需要用乙醚萃取回收,回收6次,质量损失达8.8%[35]。杂多酸的均相催化反应体系存在回收过程复杂且质量损失率高的弊端,所以降低杂多酸在水中的溶解性、将均相催化转变为非均相催化,是杂多酸催化剂的改进方向,目前主要采用金属离子或表面活性阳离子取代杂多酸质子两种方法使其非均相化。
Tian等[37]用金属离子Cs+取代杂多酸中的质子实现纤维素水解的非均相催化,制备出不溶于水的杂多酸盐催化剂CsxH3-xPW12O4,既简化了回收过程,又降低了质量损失,然而因为杂多酸中提供布朗斯特酸性的H+被Cs+取代,杂多酸H+减少,酸性降低,且取代量越多,催化活性降低幅度越大,导致催化性能降低的问题。由此从保持杂多酸强酸性和催化活性的角度来看,应该尽可能减少金属离子的取代量而保留尽可能多的质子,但CsxH3-xPW12O40回收过程的质量损失随Cs含量增加而降低,CsH2PW12O40损失量为最高的5.6%,CsPW12O40为最低的0.5%,所以从减少质量损失的角度来看,杂多酸的质子取代越多越好。因此在杂多酸金属盐催化剂中存在保持酸性和减少损失的矛盾,解决这一矛盾的思路是用具有路易斯酸性的金属离子进行替代,以路易斯酸补偿损失的布朗斯特酸。Shimizu等[34]的研究中,用路易斯酸性Sn4+完全替代H3PW12O40的质子制得Sn0.75PW12O40,其催化反应变化曲线的趋势与H3PW12O40一致且催化效果相当,如图4,催化纤维素获得的还原糖产率均为40%。通过表面活性阳离子替代杂多酸质子的非均相化方法同样因为杂多酸质子数量减少而面临降低酸性的问题,其解决思路是在反荷离子的化学结构中嫁接酸性基团来补偿损失的酸性,但目前还没有依照这种思路合成出不溶于水的杂多酸催化剂。Cheng等[38]遵循这一思路合成了一种溶于水的催化 剂,为了使催化剂具备溶解纤维素的功能,用离子液体[C4H6N2(CH2)3SO3H]+取代H3PW12O40质子,质子替代造成的酸性损失则通过嫁接在咪唑离子的—SO3H进行补偿。
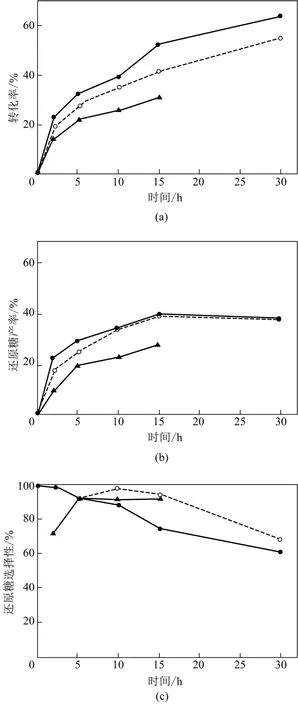
图4 H3PW12O40、Sn0.75PW12O40、H2SO4150℃下催化水解纤维素的表现[34]
纤维素与催化剂的接触困难是杂多酸非均相催化导致的问题,在杂多酸催化剂中引入具纤维素吸附能力的抗衡阳离子能够解决这一问题。Guan 等[39-40]用表面活性阳离子[C16H33N(CH3)3]+作为杂多阴离子的反荷基团,表面活性阳离子在水中形成胶束,通过与纤维素的醚键、—OH作用而相互吸附,促使分散于水中的纤维素集中在催化剂上,增加催化剂与杂多酸的接触,吸附后催化剂的酸中心能更充分发挥活性,纤维素水解最高获得还原糖产率40.2%、选择性89.1%。
强酸性是杂多酸与其他类型催化剂相比具有的明显优势,质量损失是杂多酸的应用难题,引入酸性基团维持其强酸性,同时在载体嫁接具有纤维素吸附能力的活性基团增强纤维素与催化剂的接触是杂多酸催化剂的研究方向。
6 纤维素解结晶对催化剂水解性能的影响
虽然催化剂自身物理、化学结构决定其催化性能,但纤维素的结晶结构会阻碍纤维素完全转化而影响催化效果。有研究组通过球磨处理降低纤维素结晶度,从而提高纤维素的水解效率,然而球磨处理时间长(1~2d)且能耗大,不符合经济效益。也有的研究组通过离子液体与纤维素之间形成氢键破坏纤维素结晶结构,使纤维素水解率得到提高,然而离子液体合成工艺复杂、与水解产物的分离、纯化困难等缺点限制其应用。在磺化固体酸催化剂的研究中,Pan等[24]利用CP-SO3H中—Cl与纤维素—OH的氢键有效破坏结晶结构,在节省纤维素预处理能耗的同时避免了离子液体的弊端。杂多酸催化剂的杂多酸阴离子外层结构中的氧原子可与纤维素—OH形成氢键破坏结晶结构,在Ogasawara等[41]的研究中,浸泡于高浓度Na5BW12O40溶液的纤维素结晶度明显下降,而在高浓度H5BW12O40反应体系中水解纤维素,因为结晶结构的破坏,能够获得77%的葡萄糖产率。除了利用杂多酸自身化学结构外,Cheng等[38]利用杂多酸中引入的离子液体阳离子[C4H6N2(CH2)3SO3H]+与纤维素的—OH的氢 键[42],同样能够打破纤维素结晶结构而将其溶解,获得70%纤维素转化率以及36%葡萄糖产率。以上研究证明了合成兼备解结晶能力和酸催化能力的多功能固体酸催化剂是切实可行的,所以,使固体酸催化剂具有破坏纤维素结晶结构的活性组分,通过催化剂与纤维素之间的直接作用实现纤维素结晶结构的解除是固体酸催化剂的发展趋势。
7 结 语
比较以上5种类型的催化剂,氢型沸石的孔径尺寸限制了其与纤维素的接触,催化活性难以发挥,而金属氧化物、负载金属催化剂本身酸性弱,难以提供足够的酸中心催化纤维素,催化效果不理想。磺化固体酸具有高密度的吸附中心和酸中心,吸附中心能够选择性吸附纤维素,有利于纤维素与催化剂的充分接触和葡萄糖选择性的提高,而酸中心能满足纤维素酸水解的要求。但是活性基团—SO3H的损失问题会阻碍其应用,值得重视,可通过—SO3H与催化剂内部构成的相互作用力固定—SO3H,从而减少损失。强酸性的杂多酸由于在催化纤维素水解反应中有较高的催化活性与葡萄糖选择性,受到了广泛关注,而杂多酸作为催化剂最大的弊端在于质量损失,所以在保持杂多酸强酸性的同时,通过金属替代或者固载化的方法降低质量损失是杂多酸催化剂的改进思路。
[1] Huang Y B,Fu Y. Hydrolysis of cellulose to glucose by solid acid catalysts[J].Green Chemistry,2013,15:1095-1111.
[2] Lanzafame P,Temi D M,Perathoner S,et al. Direct conversion of cellulose to glucose and valuable intermediates in mild reaction conditions over solid acid catalysts[J].Catalysis Today,2012,179:178-184.
[3] Zhang Q H,Benoit M,Vigier K D O,et al. Pretreatment of microcrystalline cellulose by ultrasounds:Effect of particle size in the heterogeneously-catalyzed hydrolysis of cellulose to glucose[J].Green Chemistry,2013,15:963-969.
[4] Hara M,Yoshida T,Takagaki A,et al. A carbon material as a strong protonic acid[J].Angewandte Chemie International Edition,2004,43:2955-2958.
[5] Shimizu K,Satsuma A. Toward a rational control of solid acid catalysis for green synthesis and biomass conversion[J].Energy Environmental Science,2011,4:3140-3153.
[6] Hara M. Biomass conversion by a solid acid catalyst[J].Energy Environmental Science,2010,3:601-607.
[7] Suganuma S,Nakajima K,Kitano M,et al. Hydrolysis of cellulose by amorphous carbon bearing SO3H,COOH,and OH groups[J]. Journal of the American Chemical Society,2008,130:12787-12793.
[8] Jiang Y J,Li X T,Cao Q,et al. Acid functionalized,highly dispersed carbonaceous spheres:An effective solid acid for hydrolysis of polysaccharides[J].Journal of Nanoparticle Research,2011,13:463-469.
[9] Liu M,Jia S Y,Gong Y Y,et al. Effective hydrolysis of cellulose into glucose over sulfonated sugar-derived carbon in an ionic liquid[J].Industrial and Engineering Chemistry Research,2013,52:8167-8173.
[10] Shen S G,Wang C Y,Cai B,et al. Heterogeneous hydrolysis of cellulose into glucose over phenolic residue-derived solid acid[J].Fuel,2013,113:644-649.
[11] Jiang Y J,Li X T,Wang X C,et al. Effective saccharification of lignocellulosic biomass over hydrolysis residue derived solid acid under microwave irradiation[J].Green Chemistry,2012,14:2162-2167.
[12] Pang J F,Wang A Q,Zheng M Y,et al. Hydrolysis of cellulose into glucose over carbons sulfonated at elevated temperatures[J].Chemistry Communication,2010,46:6935-6937.
[13] Zhang X C,Zhang Z,Wang F,et al. Lignosulfonate-based heterogeneous sulfonic acid catalyst for hydrolyzing glycosidic bonds of polysaccharides[J].Journal of Molecular Catalysis A:Chemical,2013,377 :102- 107.
[14] Suganuma S,Nakajima K,Kitano M,et al. Sp3-linked amorphous carbon with sulfonic acid groups as a heterogeneous acid catalyst[J].Chem. Sus. Chem.,2012,5:1841-1846.
[15] Onda A,Ochi T,Yanagisawa K. Selective hydrolysis of cellulose into glucose over solid acid catalysts[J]Green Chemistry,2008,10:1033-1037.
[16] Onda A,Ochi T,Yanagisawa K. Hydrolysis of cellulose selectively into glucose over sulfonated activated-carbon catalyst under hydrothermal conditions[J].Topics in Catalysis,2009,52:801-807.
[17] Guo H X,Qi X H,Li L Y,et al. Hydrolysis of cellulose over functionalized glucose-derived carbon catalyst in ionic liquid[J].Bioresource Technology,2012,116:355-359.
[18] Zhang C,Fu Z H,Liu Y C,et al. Ionic liquid-functionalized biochar sulfonic acid as a biomimetic catalyst for hydrolysis of cellulose and bamboo under microwave irradiation[J].Green Chemistry,2012,14:1928-1934.
[19] Lai D M,Deng L,Li J,et al Hydrolysis of cellulose into glucose by magnetic solid acid[J].Chem. Sus. Chem.,2011,4:55-58.
[20] 王华瑜,张长斌,贺泓,等. 磁性碳基磺酸化固体酸催化剂的制备及其催化水解纤维素[J].物理化学学报,2010,26(7):1873-1878.
[21] Zhang C B,Wang H Y,Liu F D,et al. Magnetic core-shell Fe3O4@C-SO3H nanoparticle catalyst for hydrolysis of cellulose[J].Cellulose,2013,20:127-134.
[22] Akiyama G,Matsuda R,Sato H,et al. Cellulose hydrolysis by a new porous coordination polymer decorated with sulfonic acid functional groups[J].Advanced Materials,2011,23:3294-3297.
[23] Liu F J,Zheng A M,Noshadi I,et al. Design and synthesis of hydrophobic and stable mesoporous polymeric solid acid with ultra strong acid strength and excellent catalytic activities for biomass transformation[J].Applied Catalysis B:Environmental,2013,136- 137:193- 201.
[24] Shuai L,Pan X J. Hydrolysis of cellulose by cellulase-mimetic solid catalyst[J].Energy Environmental Science,2012,5:6889-6894.
[25] Karaki M,Karout A,Toufaily J,et al. Synthesis and characterization of acidic ordered mesoporous organosilica SBA-15:Application to the hydrolysis of cellobioseand insight into the stability of the acidic functions[J].Journal of Catalysis,2013,305:204-216.
[26] Fan G Z,Liao C J,Fang T,et al. Hydrolysis of cellulose catalyzed by sulfonated poly(styrene-co-divinylbenzene) in the ionic liquid 1-n-butyl-3-methylimidazolium bromide[J].FuelProcessing Technology,2013,116:142-148.
[27] Zhou L P,Liu Z,Shi M T,et al. Sulfonated hierarchical H-USY zeolite for efficient hydrolysis of hemicellulose/cellulose[J].Carbohydrate Polymer,2013,98:146- 151.
[28] Zhou L P,Shi M T,Cai Q Y,et al. Hydrolysis of hemicellulose catalyzed by hierarchical H-USY zeolites. The role of acidity and pore structure[J].Microporous and Mesoporous Materials,2013,169:54-59.
[29] Cai H L,Li C Z,Wang A Q,et al. Zeolite-promoted hydrolysis of cellulose in ionic liquid,insight into the mutual behavior of zeolite,cellulose and ionic liquid[J].Applied Catalysis B:Chemical,2012,123- 124:333- 338.
[30] Takagaki A,Tagusagawa C,Domen K. Glucose production from saccharides using layered transition metal oxide and exfoliated nanosheets as a water-tolerant solid acid catalyst[J].Chemistry Communication,2008,42:5363.
[31] 唐元,何杰,张忠良,等. 层状铌基复合氧化物固体酸及其醋酸正丁醇酯化的催化活性[J]. 广东化工,2011(5):67-68.
[32] Komanoya T,Kobayashi H,Hara K,et al. Catalysis and characterization of carbon-supported ruthenium for cellulose hydrolysis[J]. Applied Catalysis A:General,2011,407:188- 194.
[33] Kobayashi H,Ito Y,Komanoya T,et al. Synthesis of sugar alcohols by hydrolytic hydrogenation of cellulose over supported metal catalysts[J].Green Chemistry,2011,13:326-333.
[34] Shimizu K,Furukawa H,Kobayashi N,et al. Effects of Bronsted and Lewis acidities on activity and selectivity of heteropolyacid-based catalysts for hydrolysis of cellobiose and cellulose[J].Green Chemistry,2009,11:1627-1632.
[35] Tian J,Wang J H,Zhao S,et al. Hydrolysis of cellulose by the heteropoly acid H3PW12O40[J].Cellulose,2010,17:587-594.
[36] Yang J,Janik M J,Ma D,et al. Location,acid strength,and mobility of theacidic protons in Keggin 12-H3PW12O40:A combined solid-state NMR spectroscopy and DFT quantum chemical calculation study[J].Journal of American Chemical Society,2005,127:18274-18280.
[37] Tian J,Fan C Y,Cheng M X,et al. Hydrolysis of cellulose over CsxH3-xPW12O40(x= 1—3) heteropoly acid catalysts[J].Chemical Engineering and Technology,2011,34(3):482-486.
[38] Sun Z,Cheng M X,Li H C,et al. One-pot depolymerization of cellulose into glucose and levulinic acid by heteropolyacid ionic liquid catalysis[J].RSC Advances,2012,2:9058-9065.
[39] Cheng M X,Shi T,Guan H Y,et al. Clean production of glucose from polysaccharides using a micellar heteropolyacid as a heterogeneous catalyst[J].Applied Catalysis B:Chemical,2011,107:104-109.
[40] Cheng M X,Shi T,Wang S T,et al. Fabrication of micellar heteropolyacid catalysts for clean production of monosaccharides from polysaccharides[J].Catalysis Communications,2011,12:1483-1487.
[41] Ogasawara Y,Itagaki S,Yamaguchi K,et al. Saccharification of natural lignocellulose biomass and polysaccharides by highly negatively charged heteropolyacids in concentrated aqueous solution[J].Chem. Sus. Chem.,2011,4:519-525.
[42] Liu D T,Xia K F,Cai W H,et al. Investigations about dissolution of cellulose in the 1-allyl-3-alkylimidazolium chloride ionic liquids[J].Carbohydrate Polymer,2012,87:1058-1064.
