反相离子对HPLC法同时测定盐酸二甲双胍中双氰胺和三聚氰胺的含量
2014-08-04王丽娟刘军英刘惠涛
王丽娟,刘军英,刘惠涛
(烟台大学化学化工学院,山东 烟台 264005)
盐酸二甲双胍(Metformin Hydrochloride)又名1,1-二甲基双胍盐酸盐,为双胍类口服降血糖药,治疗非胰岛素依赖型糖尿病[1],主要作用于胰岛外组织,抑制肠吸收葡萄糖[2].双氰胺(Cyanoguanidine,CGD)和三聚氰胺(Melamine,MEL)都是盐酸二甲双胍的合成中间体(图 1),毒性较大,必须严格控制其在药品中的含量,保证患者用药安全、有效,目前规定其限量分别为0.02%和0.1%[3].
双氰胺和三聚氰胺的检测方法包括气相色谱[4]、高效液相色谱[3,5-10]、薄层层析[11]、毛细管电泳[12-13]和表面增强拉曼散射光谱法[14]等.但采用简单的反相HPLC法同时测定盐酸二甲双胍中的双氰胺和三聚氰胺含量的方法还未见报道.本文使用C18反相色谱柱,通过对离子对试剂的种类、配比、pH值等因素的系统考察,建立一种测定盐酸二甲双胍制剂中的杂质双氰胺和三聚氰胺的离子对色谱法.

图1 双氰胺和三聚氰胺的结构式
1 实验部分
1.1 仪器与试剂
Waters 1525型高效液相色谱仪,配有Waters 2487型紫外检测器,美国Waters 公司;pH/Lon 510型酸度计,新加坡Crison公司,型号pH 5500,精度0.01个pH单位;双光束紫外可见分光光度计,型号TU-1901,北京普析通用仪器有限公司;超声波清洗仪,型号SK3300HP,上海科导超声有限公司;分析天平,型号AL-204,梅特勒-托利多仪器(上海)有限公司.
双氰胺对照品(中国食品药品检定研究院),三聚氰胺对照品(Dr. Ehrenstorfer股份有限公司(德国)),庚烷磺酸钠、戊烷磺酸钠、乙腈、甲醇均为色谱纯,柠檬酸为分析纯,盐酸二甲双胍片2批,样品① 批号:H11021518,样品② 批号:H20060614.实验用水均为超纯水.
1.2 实验方法
1.2.1 色谱条件 色谱柱:Hypersil ODS C18反相色谱柱 (5 μm; 250×4.6 mm);流动相:乙腈-离子对试剂水溶液(10 mmol/L戊烷磺酸钠+10 mmol/L柠檬酸,pH值为3.0,体积比10∶90);流速:1.0 mL/min;检测波长:217 nm;进样量:20 μL;柱温:25 ℃.
1.2.2 溶液的制备 双氰胺对照品溶液:取双氰胺对照品约5 mg,精密称定,置5 mL容量瓶中,加入流动相溶解并稀释至刻度,得双氰胺对照品储备液.
三聚氰胺对照品溶液:取三聚氰胺对照品约5 mg,精密称定,置10 mL容量瓶中,加入流动相溶解并稀释至刻度,得三聚氰胺对照品储备液.
混合标准溶液:分别精密量取双氰胺对照品储备液和三聚氰胺对照品储备液各1.0 mL,置100 mL容量瓶中,加入流动相溶解并稀释至刻度,制成每1 mL中约含双氰胺10 μg和三聚氰胺5 μg的混合标准溶液.
供试品溶液:取盐酸二甲双胍片样品① 、②各3片,精密称定,研细,精密称取适量(约相当于盐酸二甲双胍500 mg),置100 mL容量瓶中,加超纯水20 mL,超声处理15 min后流动相稀释至刻度,摇匀,过滤.所用溶液分析前均经0.45 μm滤膜过滤.
2 结果与讨论
2.1 色谱条件的选择
2.1.1 检测波长的确定 经过紫外双光束分光光度计扫描(190~400 nm),得到双氰胺的最大吸收波长是217 nm,三聚氰胺的最大吸收波长是204 nm.而国标中三聚氰胺的检测波长为240 nm,因此分别测定217 nm和240 nm波长下的色谱,离子对试剂为戊烷磺酸钠,对乙腈的体积比为90∶10,试验浓度为10 mmol/L(含10 mmol/L柠檬酸,pH值为3.0),结果显示在240 nm下CGD 和MLN的吸收峰均较弱,因此选择217 nm为2种物质的最佳检测波长.
2.1.2 离子对试剂种类的选择 选用的离子对试剂为戊烷磺酸钠和庚烷磺酸钠,试验浓度为10 mmol/L(含10 mmol/L柠檬酸,pH值为3.0),对乙腈的体积比为90∶10.结果显示,使用戊烷磺酸钠最为离子对试剂的保留时间更短,且分离良好.综合考虑溶剂峰与双氰胺峰的分离度以及保留时间的差异,最终选择戊烷磺酸钠作为离子对试剂.
2.1.3 离子对试剂pH值的选择 以戊烷磺酸钠为离子对试剂,浓度为10 mmol/L,与乙腈体积比为90∶10,分别考察了pH值为2.5、3.0、3.5、4.0不同条件下的分离情况.结果显示,在不同pH值条件下,双氰胺的保留时间均变化不显著,而三聚氰胺的保留时间随pH值增大而延长,pH值为2.5和4.0时双氰胺与溶剂峰不能分离,当流动相pH值为3.0时双氰胺和三聚氰胺具有最佳分离和最短分析时间,因此选择离子对试剂溶液的pH值为3.0.
2.1.4 流动相比例的选择 采用10 mmol/L的戊烷磺酸钠(pH值为3.0),分别考察了流动相中离子对试剂与乙腈的体积比为85∶15、90∶10、95∶5的不同实验条件下,双氰胺和三聚氰胺的分离度.结果显示,流动相比例为90∶10时双氰胺和三聚氰胺具有最佳分离,因此选择流动相的比例为90∶10.
综上所述,最优分离条件为乙腈-离子对试剂水溶液(10 mmol/L戊烷磺酸钠+10 mmol/L柠檬酸,pH值为3.0,体积比10∶90).
2.2 方法验证
2.2.1 线性 分别配制双氰胺和三聚氰胺一系列不同浓度的混合标准溶液,在最优色谱条件下进行测定,以峰面积对组分浓度分别作图,在0.02~20 μg/mL浓度范围内得到双氰胺回归方程为A双氰胺=229 028c双氰胺+528.24,R=0.999 9;在0.02~50 μg/mL浓度范围内,三聚氰胺回归方程是A三聚氰胺=159 033c三聚氰胺+16 650,R=0.999 9.表明在试验浓度范围内峰面积与浓度线性良好.
2.2.2 检出限和定量限 取对照品溶液稀释到合适浓度,进样,以信噪比的3倍和10倍计算检出限和定量限.双氰胺的检出限为4.0 ng/mL,定量限为15.0 ng/mL;三聚氰胺的检出限为6.0 ng/mL,定量限为20.0 ng/mL.
2.2.3 精密度 取2种物质的标准品溶液日内和日间连续进样5次,计算日内精密度和日间精密度,结果如表1所示.
2.3 样品测定
在上述优化的色谱条件下,对盐酸二甲双胍片中的三聚氰胺和双氰胺的杂质含量进行测定,结果如图2中b、c所示.2种批号的药品中均未检出三聚氰胺和双氰胺.

表1 HPLC测定CGD和MLN的精密度(n=5)
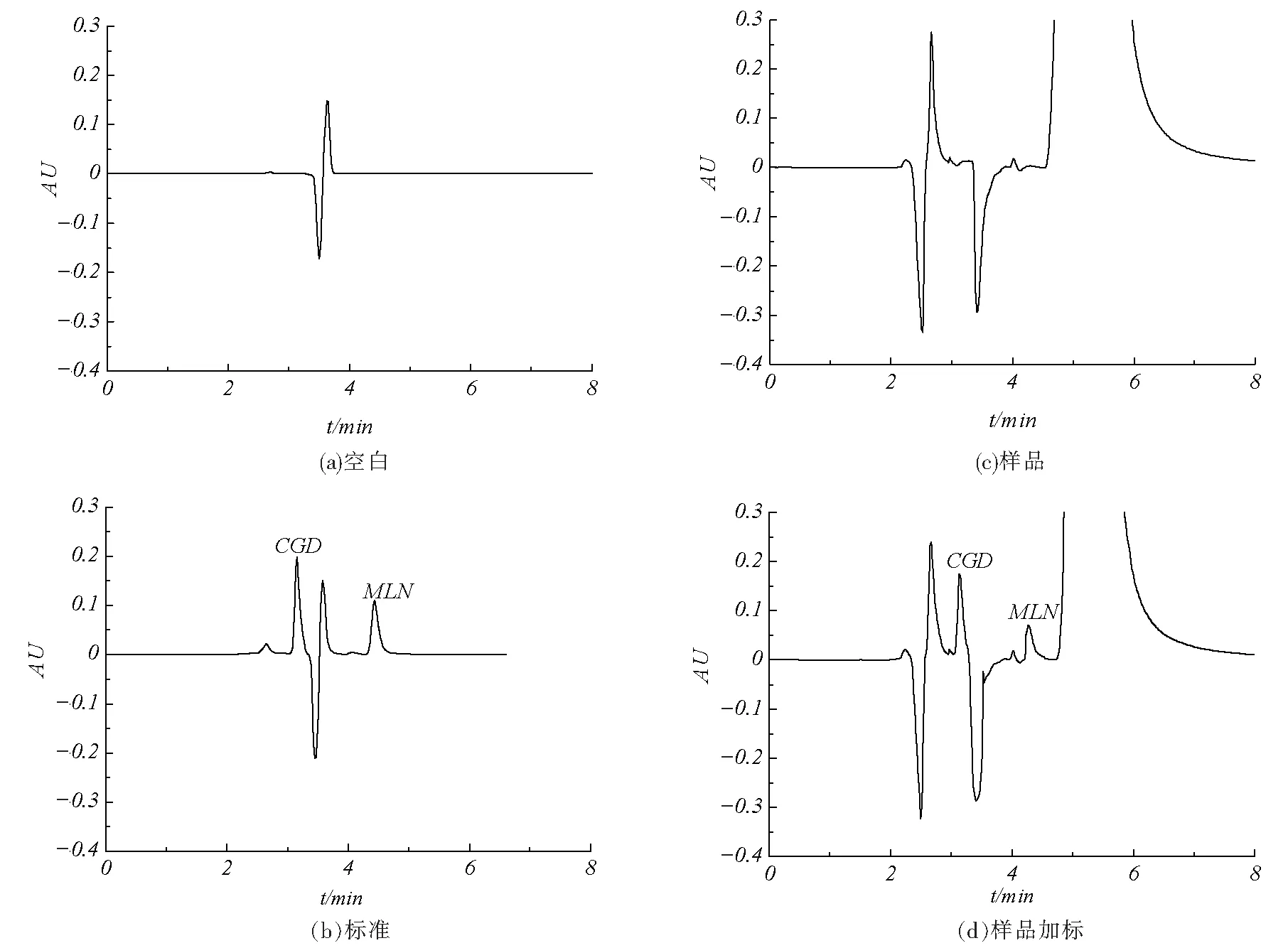
图2 空白、标准、样品及样品加标色谱图
2.4 回收率
在2种样品中加入已知浓度的2种标准物质,按照样品处理步骤处理样品后进样.根据色谱图峰面积计算2种物质的回收率,结果如表2所示.从表中可以看出,双氰胺的平均回收率是97.6%,三聚氰胺的平均回收率是96.5%.说明HPLC法测定样品中双氰胺和三聚氰胺回收率良好.
3 方法讨论
双氰胺和三聚氰胺均为极性化合物,很难在常用的C18柱上保留.因此,文献中测定盐酸二甲双胍药品中的双氰胺和三聚氰胺所使用的方法为亲水相互作用色谱法(Hydrophilic Interaction Liquid Chromatography,HILIC).但此方法需要使用价格较为昂贵的特定色谱柱.而本研究中使用常见的ODS C18反相色谱柱,加入离子对试剂,2种杂质在5 min内分离完全,保留时间更短,且检出限和定量限均低于上述方法,是一种简单经济的有效测定方法.
4 结 论
本研究建立了一种高效快速的HPLC方法同时测定盐酸二甲双胍药品中的双氰胺和三聚氰胺,2种杂质在5 min内分离完全.该方法分离效率高,分析时间短,灵敏度好,样品处理简单快速,是盐酸二甲双胍药品中杂质检测的一种有效方法.

表2 样品中CGD和MLN的回收率的测定(n=3)
注:—为未检出.
参考文献:
[1] 杨亚军. HPLC法测定盐酸二甲双胍缓释片中杂质双氰胺含量[J]. 沈阳药科大学学报,2004,21(6): 436-437,457.
[2] 吴莉,石蓓佳,王玉,等. 反相离子对HPLC法测定盐酸二甲双胍中双氰胺的含量[J]. 中国生化药物杂志,2009,30(2): 110-113.
[3] Ali M S. Simultaneous determination of metformin hydrochloride,cyanoguanidine and melamine in tablets by mixed-mode HILIC[J]. Chromatographia,2008,67(7-8): 517-525.
[4] Inoue T,Ishiwata H,Yoshihira K,et al. High-performance liquid chromatographic determination of melamine extracted from cups made of melamine resin[J]. J Chromatogr A,1985,346: 450-452.
[5] Ehling S,Tefera S,Ho I P,et al. High-performance liquid chromatographic method for the simultaneous detection of the adulteration of cereal flours with melamine and related triazine by-products ammeline,ammelide,and cyanuric acid[J]. Food Addit Contam Part A,2007,24(12): 1319-1325.
[6] Muniz-Valencia R,Ceballos-Magana S G,Rosales-Martinez D,et al. Method development and validation for melamine and its derivatives in rice concentrates by liquid chromatography: Application to animal feed samples[J]. Anal Bioanal Chem,2008,392(3): 523-531.
[7] Sancho J V,Ibanez M,Grimalt S,et al. Residue determination of cyromazine and its metabolite melamine in chard samples by ion-pair liquid chromatography coupled to electrospray tandem mass spectrometry[J]. Anal Chim Acta,2005,530(2): 237-243.
[8] Filigenzi M S,Tor E R,Poppenga R H,et al. The determination of melamine in muscle tissue by liquid chromatography/tandem mass spectrometry[J]. Rapid Commun Mass Spectrom,2007,21(24): 4027-4032.
[9] 牟玲. HPLC测定复方盐酸二甲双胍片中有关物质双氰胺[J]. 中国药品标准,2006,7(4): 44-46.
[10] 孙桂荣,卢华伟,刘爱云,等. 高效液相色谱法测定盐酸二甲双胍缓释片的有关物质[J].中国药业,2004,13(10): 34-35.
[11] Broszat M,Bramer R,Spangenberg B,et al. A new method for quantification of melamine in milk by absorption diode-array thin-layer chromatography[J]. J Planar Chromatogr-Mod TLC,2008,21(6): 469-470.
[12] Yan Na,Zhou Lei,Zhu Zaifang,et al. Determination of melamine in diary products,fish feed,and fish by capillary zone electrophoresis with diode array detection[J]. J Agric Food Chem,2009,57(3): 807-811.
[13] Meng Liang,Shen Guijun,Hou Xiaolan,et al. Determination of melamine in food by SPE and CZE with UV detection[J]. Chromatographia,2009,70(5-6): 991-994.
[14] Okazaki S,Gonmori M H K,Suzuki O,et al. Rapid nondestructive screening for melamine in dried milk by Raman spectroscopy[J]. Forensic Toxicol,2009,27(2): 94-97.
