柱后衍生高效液相色谱法测定虾中14种磺胺类药物残留量
2014-08-03黄冬梅黄宣运顾润润惠芸华田良良于慧娟
黄冬梅, 黄宣运, 顾润润, 惠芸华, 田良良, 冯 兵, 张 璇, 于慧娟
(中国水产科学研究院东海水产研究所,上海200090)
磺胺类(sulfonamides,简称SAs)是指具有氨基苯磺酰胺结构的一类药物的总称。磺胺类药物对革兰氏阳性菌和一些革兰氏阴性菌有显著的治疗作用,能治疗水产动物的细菌性疾病,因此在水产养殖中使用较为普遍。过量使用磺胺类药物会导致水产品中存在药物残留,人们食用这些水产品后就可能造成体内药物蓄积,对人体健康产生危害。SAs能破坏人的造血系统,造成溶血性贫血症、粒细胞缺乏症和血小板减少症等,同时具有潜在的致畸、致癌或致突变作用[1]。因此磺胺类药物在生物体中的残留情况日益受到关注。日本将磺胺喹噁啉、磺胺二甲基嘧啶、磺胺-6-甲氧嘧啶和磺胺嘧啶列为动物性食品重点监控的药物,规定磺胺二甲基嘧啶的最大残留限量为10 μg/kg。美国规定泌乳牛禁用磺胺二甲基嘧啶等。我国农业部第235号公告规定所有动物食品中磺胺类药物总量不超过100 μg/kg的最大残留限量值[2]。近几年我国加强了对养殖水产品中磺胺类药物的抽检力度,以确保水产品的质量安全。
国内外磺胺类药物的残留检测方法包括酶联免疫 法[3-5]、安 培 检 测 法[6]、高 效 液 相 色 谱 法(HPLC)[7-14]、液相色谱-串联质谱法[15-18]等。目前较为常用的是高效液相色谱-荧光检测法和紫外检测法,以及液相色谱-串联质谱法。张小军等[19]利用液相色谱-荧光检测法测定水产品中4种磺胺类药物,使用荧光胺作为柱前衍生试剂,检出限(LOD)仅为 10 μg/kg。Sílvia 等[20]也采用荧光胺柱前衍生法对动物饲料中的8种磺胺进行测定,荧光反应于暗处室温下反应2 h,不同动物饲料中的LOD为10~100 μg/kg。而配有紫外检测器的液相色谱法的检测灵敏度更低,卢坤等[21]采用高效液相色谱-紫外检测法测定蜂蜜中5类抗生素,磺胺类药物的定量限(LOQ)为33.27 ~40.13 μg/kg,同时液相色谱-紫外检测法通常采用选择性较低的190 nm或270 nm的紫外波长检测,会有低吸收波长的杂质化合物干扰目标化合物的分离,从而影响定量结果的准确性。液相色谱-串联质谱法灵敏度较高,张海琪等[22]利用液相色谱-串联质谱法测定大黄鱼中20种磺胺类药物残留,LOQ均能达到5.0 μg/kg,但是液相色谱-串联质谱仪较昂贵,使用成本高。
本研究建立了水产品中磺胺类残留检测的在线柱后衍生液相色谱法,磺胺类药物经色谱柱分离后,在柱后衍生系统中与荧光胺(fluorescamine)反应,相比柱前衍生提高了衍生反应速度,缩短了检测时间,反应生成具有专一荧光特性的化合物,用荧光检测器定性定量分析,可明显消除杂质干扰;同时本文对衍生反应参数进行优化,提高了检测响应值,使14种磺胺类药物的定量限达到1.0~5.0 μg/kg,与液相色谱-串联质谱法的灵敏度接近。该方法的建立可以提供灵敏度高、结果准确可靠的虾中磺胺类药物的多残留测定。
1 实验部分
1.1 仪器、试剂与样品
1100高效液相色谱仪(美国Agilent公司),配有荧光检测器,PCX型柱后衍生系统(美国Pickering Vector公司);CF16RXⅡ型离心机(日本Hitachi公司);旋转蒸发仪(上海亚荣生化仪器厂);漩涡混合器(德国IKA公司);超声波清洗器(美国Branson公司);Milli-Q超纯水装置(Millipore公司)。
磺胺类药物标准品:磺胺(sulfanilamide,SN)、磺胺嘧啶(sulfadiazine,SD)、磺胺噻唑(sulfathiazole,ST)、磺胺甲基嘧啶(sulfamerazine,SM1)、磺胺-5-甲氧嘧啶(sulfameter,SMD)、磺胺二甲基嘧啶(sulfamethazine,SM2)、磺胺甲氧哒嗪(sulfamethoxypyridazine,SMP)、磺胺氯哒嗪 (sulfachlorpyridazine,SCP)、磺胺-6-甲氧嘧啶(sulfamonomethoxine,SMM,纯度 98.5%)、磺胺甲噁唑(sulfamethoxazole,SMZ)、磺胺多辛(sulfadoxine,SDM')、磺 胺 二 甲 异 噁 唑 (sulfisoxazole,SXZ)、磺胺二甲氧哒嗪(sulfadimethoxine,SDM)、磺胺喹噁啉(sulfaquinoxaline,SQ,纯度98.0%)、磺胺吡啶(sulfapyridine,SP,内标)均购自德国Dr.Ehrenstorfer公司(未标注纯度的标准品纯度均≥99.0%,);甲醇、乙腈、正己烷、乙酸乙酯为色谱级试剂(美国Tedia公司);荧光胺(纯度≥98.0%,美国Sigma公司);乙酸、盐酸、乙酸钠(分析纯,国药化学试剂公司);微孔滤膜(0.45 μm,上海安谱科学仪器公司);超纯水(18.2 MΩ·cm)。
混合标准溶液:准确称取各磺胺类药物标准品,分别用甲醇溶解,配制成以下质量浓度的标准溶液,磺胺 8 mg/L、磺胺嘧啶 16 mg/L、磺胺噻唑 16 mg/L、磺胺甲基嘧啶16 mg/L、磺胺-5-甲氧嘧啶40 mg/L、磺胺二甲基嘧啶16 mg/L、磺胺甲氧哒嗪40 mg/L、磺胺氯哒嗪40 mg/L、磺胺-6-甲氧嘧啶40 mg/L、磺胺甲噁唑40 mg/L、磺胺多辛40 mg/L、磺胺二甲异噁唑40 mg/L、磺胺二甲氧哒嗪40 mg/L、磺胺喹噁啉40 mg/L。分别取标准溶液5 mL,用甲醇稀释并定容至100 mL。
内标标准溶液:准确称取磺胺吡啶标准品8 mg,用甲醇溶解定容至100 mL。溶液浓度为80 mg/L。取2.5 mL,用乙酸乙酯稀释并定容至100 mL,溶液质量浓度2 mg/L。
2%乙酸溶液:量取20 mL乙酸,加水至1 000 mL;0.2 g/L荧光胺溶液:称取0.04 g荧光胺,加入100 mL乙腈溶解,再加入20 mL甲醇和80 mL乙酸溶液混匀,保存在棕色瓶中,现用现配;2 mol/L盐酸溶液:量取180 mL盐酸,加水至1 000 mL;3.5 mol/L乙酸钠溶液:称取28.7 g无水乙酸钠,加水溶解并稀释至100 mL;乙腈、甲醇、乙酸钠混合溶液:准确移取20 mL乙酸钠溶液、5 mL乙腈和5 mL甲醇混合。
凡纳滨对虾、青虾、罗氏沼虾为购自上海市农贸市场的养殖水产品。
1.2 仪器条件
1.2.1 液相色谱条件
色谱柱:Agilent SB-C18柱(250 mm ×4.6 mm,5 μm);柱流速:0.6 mL/min;进样量:50 μL;柱温:40℃;激发波长:450 nm;发射波长:495 nm;流动相:A为乙腈,B为甲醇,C为2%的乙酸溶液;梯度洗脱程序为0~15 min,5%A,10%B;15~30 min,5%A~20%A,10%B;30~40 min,20%A,10%B;40~45 min,20%A~5%A,10%B;45~55 min,5%A,10%B。
1.2.2 柱后衍生系统条件
荧光胺衍生试剂质量浓度:0.2 g/L;衍生试剂流速:0.15 mL/min;反应器温度:50℃。
1.3 样品前处理
提取:虾去头、去壳、去肠腺,取肌肉部分绞碎。称取(10.00±0.05)g样品于50 mL离心管中,加入磺胺吡啶内标工作液100 μL,加入25 mL乙酸乙酯,漩涡振荡提取5 min,超声提取10 min,4 000 r/min离心5 min,将提取溶液转入鸡心瓶中;残渣按上述方法再提取一次;合并提取液,于40℃减压浓缩至约1 mL。
净化:将鸡心瓶中的液体转移至15 mL的离心管中,用4 mL乙酸乙酯洗鸡心瓶,溶液合并至离心管中,在离心管中精确加入2.00 mL盐酸溶液,漩涡混合1 min,3 000 r/min离心5 min,去除上层溶液。在离心管中加入5 mL正己烷,漩涡混合1 min,3 000 r/min离心5 min,移去正己烷层。将离心管中溶液过0.45 μm 水相滤膜,取200 μL滤液于进样瓶中,加入300 μL乙腈、甲醇、乙酸钠混合溶液,混匀1 min,供高效液相色谱测定。
2 结果与讨论
2.1 方法条件的研究
2.1.1 内标物的选择
内标法校正可消除由于测试条件的波动而对分析结果产生的影响。在痕量物质检测中内标法的使用较为普遍。选择内标物时要求在待测试样中不存在该化合物,同时内标物的化学性质应与待测组分的性质相近,并且在色谱图上能彼此分开。磺胺吡啶同属于磺胺类药物,在虾类养殖中不使用该种物质,在图1中可看出能与待测组分完全分开,不干扰待测物的出峰,因此将磺胺吡啶确定为本方法的内标物。
2.1.2 前处理方法的确定
本实验分别采用乙腈和乙酸乙酯作为提取剂,比较提取效果。结果发现乙腈对磺胺等几种药物的提取效率低;此外,由于乙腈与水互溶,提取时将水溶性杂质一并提取出来,不仅在旋转蒸发浓缩时难以蒸发,而且提取液中杂质较多影响下一步的净化;因此采用与水不互溶的乙酸乙酯作为提取剂。不同提取试剂对回收率的影响见表1。
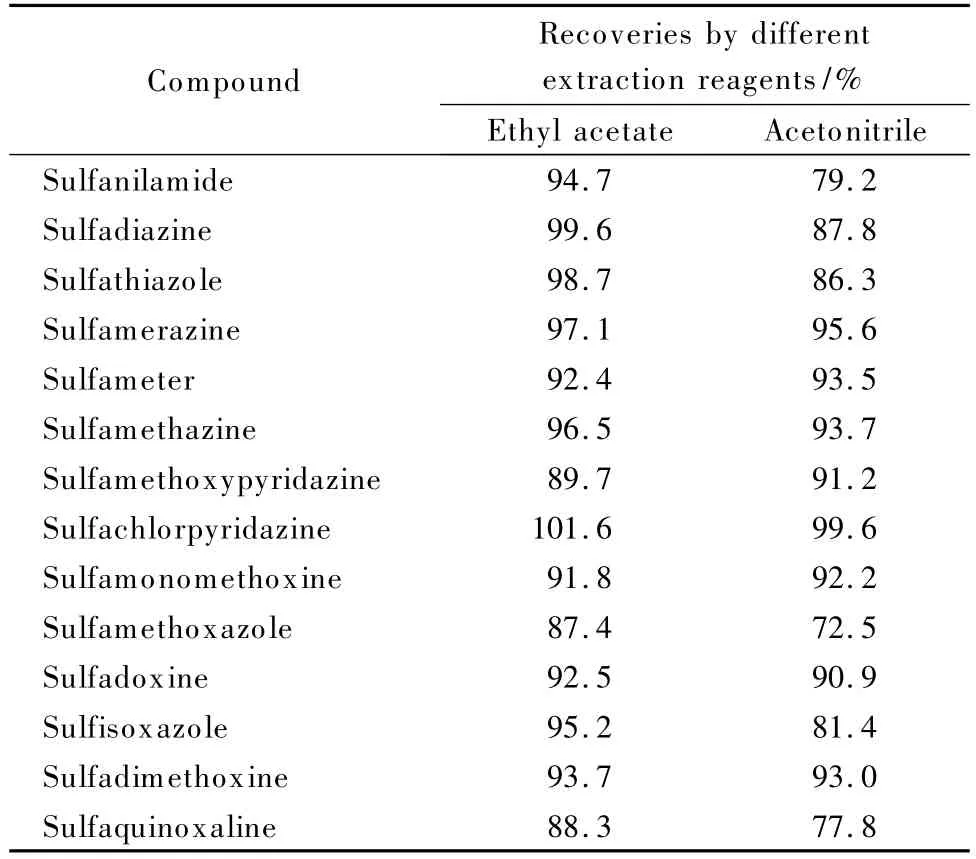
表1 不同提取试剂对磺胺回收率的影响(n=5)Table 1 Influences of different extraction reagents on recoveries of the sulfonamides(n=5)
常用的样品净化方法为液液萃取净化和固相萃取柱净化等。磺胺类药物结构中有磺酸基和氨基,可以采用磺酸基阳离子固相萃取柱净化的方法。刘海新等[14]使用磺酸基阳离子固相萃取小柱(500 mg/3 mL)进行净化,将提取液蒸发浓缩后与5 mL乙腈-二氯甲烷(4∶6,v/v)混匀,过柱后用乙腈-二氯甲烷清洗蒸发瓶并过固相萃取柱,再用5 mL乙腈、3 mL水洗涤小柱,氮气吹干后用乙酸乙酯-甲醇-氨水(50∶45∶5,v/v/v)将磺胺类药物洗脱下来,收集洗脱液,氮气吹干。过柱净化时间较长,操作步骤复杂。因此本工作利用磺胺类药物易溶于盐酸溶液的性质,采用液液萃取的方式,在反萃取时将虾中的脂溶性杂质保留在上层乙酸乙酯和正己烷层中去除,从而达到净化下层水相的目的。
磺胺类物质难溶于水,可溶于甲醇、乙醇等有机试剂。其药物分子中既有芳香第一胺(呈弱碱性),又有磺酰氨基(显弱酸性),因此磺胺类药物呈酸碱两性,可与酸或碱成盐而溶于水。因涉及下一步骤的去脂,必须将磺胺类药物保留在水相中才能用正己烷去脂,因此选择2 mol/L的盐酸溶液将磺胺类物质从乙酸乙酯层中反萃取出来,将磺胺类物质保留在盐酸溶液中。
磺胺类药物的上机溶液通常选择甲醇水溶液、乙腈水溶液或流动相,由于本方法中过水相滤膜后的滤液为2 mol/L的盐酸溶液,酸性较强,必须加入弱碱性的乙酸钠溶液降低溶液的酸性,否则上机液酸性过强会影响色谱峰峰形,给定量带来误差,同时滤液中加入有机相乙腈和甲醇,与流动相保持一致,可以改善峰形,使保留时间更稳定。
2.1.3 仪器条件的优化
选择HPLC较为常用的C18柱作为色谱柱,结果发现14种磺胺类化合物在Agilent SB-C18色谱柱上能获得理想的分离效果。
磺胺类化合物极性差异较大,采用等度洗脱很难将多组分磺胺类化合物同时分离,而采用梯度洗脱方式可以较好地解决这一问题。在梯度洗脱初始阶段以低比例有机相作为流动相,将极性较强的组分先洗脱出来,随后逐渐增加有机相比例,使非极性强的组分逐一流出色谱柱,从而实现了磺胺类药物的有效分离。同时根据磺胺类化合物具有弱碱性的性质,在流动相中加入一定的酸性物质(2%乙酸),会促进磺胺类药物的电离,有助于色谱柱内硅胶硅醇基的质子化,消除硅醇基与磺胺之间的相互作用,减少峰拖尾,改善了磺胺的色谱峰形和分离效果。磺胺类药物在阴性虾样品中的加标色谱图见图1。
2.1.4 柱后衍生系统参数的选择
柱后衍生系统的参数条件设置决定磺胺类化合物的仪器响应值,对方法的灵敏度影响很大,因此必须对参数进行优化。实验分别优化了荧光胺溶液浓度、试剂流速和反应温度这3个参数。14种磺胺类药物的变化趋势基本一致,将其中8种磺胺的结果绘成曲线,结果见图2。磺胺类化合物的面积响应值随着荧光胺溶液浓度的增加而增长,但是荧光胺浓度过高时会有析出,沉积在仪器上而对仪器造成损伤,因此确定0.2 g/L为本实验荧光胺溶液的质量浓度,在此浓度下化合物的灵敏度已能满足检测要求。当衍生化试剂流速达到0.15 mL/min时,峰面积达到最高。在反应温度达到40~50℃时,反应趋稳,响应值变化不大,因此将50℃确定为实验温度。
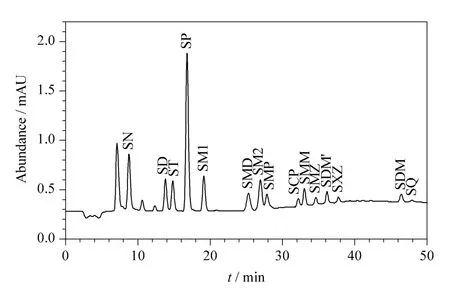
图1 空白虾样品中添加5 μg/kg磺胺类药物的色谱图Fig.1 Chromatogram of the sulfonamides spiked in a blank shrimp sample

图2 荧光胺溶液的(a)浓度、(b)流速和(c)反应温度对磺胺类药物峰面积的影响Fig.2 Influence of(a)mass concentration,(b)flow rate of fluorescamine solution and(c)reaction temperature on the peak area of the sulfonamides
2.2 方法有效性研究
2.2.1 方法线性范围及检出限、定量限
本实验的校准曲线采用基质标样添加法绘制,即在10 g空白虾样品中添加磺胺类混合标准溶液0.025、0.1、0.25、0.5、1 mL 和内标使用液50 μL,按1.3节方法操作,取50 μL注入高效液相色谱仪中,按以上仪器条件进行分析,以磺胺类化合物和内标物的峰面积比值为纵坐标Y,对应待测物的质量浓度为横坐标X,得到校准曲线,14种磺胺类药物在5~200 μg/L的范围内具有良好的线性关系。
在空白虾样品中分别添加磺胺类药物混合标准溶液,以3倍信噪比(S/N≥3)对应的质量浓度为方法的检出限,以10倍信噪比(S/N≥10)对应的质量浓度为方法的定量限。磺胺类药物的线性回归方程以及检出限、定量限见表2。
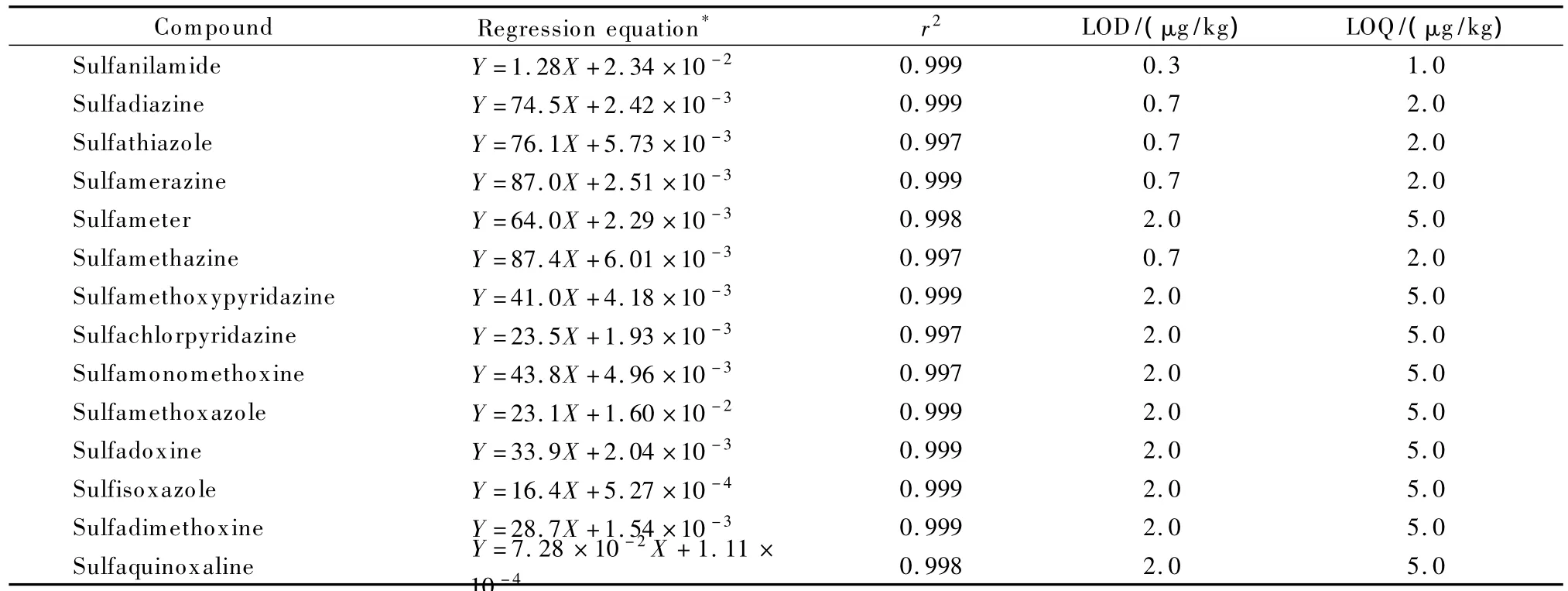
表2 14种磺胺类药物的线性回归方程、相关系数、检出限及定量限Table 2 Linear regression equations,correlation coefficients(r2),LODs and LOQs of the 14 sulfonamides
2.2.2 方法的回收率与精密度
在空白虾样品中分别添加3个水平的磺胺类药物标准溶液进行回收率试验,每个加标水平做6个平行样。测定结果显示,平均添加回收率为77.8%~103.6%,相对标准偏差为2.9% ~9.1%,具体数据见表3。
2.3 实际样品测定
应用本方法对购于上海市农贸市场的凡纳滨对虾、青虾和罗氏沼虾样品进行了14种磺胺类药物的检测,60个样品中均未检出磺胺、磺胺嘧啶、磺胺噻唑、磺胺甲基嘧啶、磺胺-5-甲氧嘧啶、磺胺甲氧哒嗪、磺胺氯哒嗪、磺胺-6-甲氧嘧啶、磺胺多辛、磺胺二甲异噁唑、磺胺二甲氧哒嗪、磺胺喹噁啉。但是磺胺二甲基嘧啶和磺胺甲噁唑有检出:在2个凡纳滨对虾样品中检出了磺胺二甲基嘧啶,含量分别为16.4 μg/kg和12.1 μg/kg;在1个青虾样品中检出了磺胺甲噁唑,含量为42.5 μg/kg;但这3个样品中磺胺类药物的总量均在国家规定的100 μg/kg的限量标准范围之内,为合格样品。
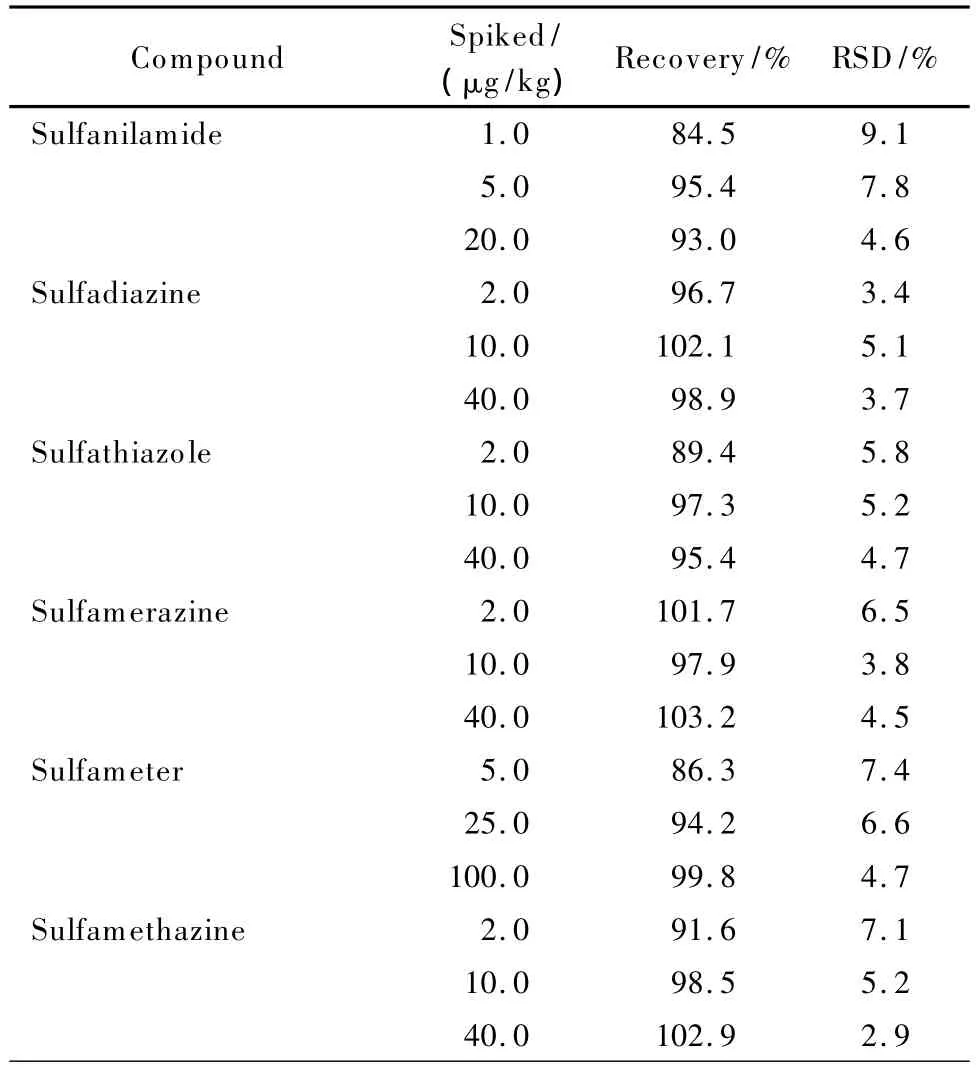
表3 虾空白样品中添加磺胺类药物的平均回收率及精密度(n=6)Table 3 Recoveries and precisions of the SAs in black shrimp samples(n=6)

表3 (续)Table 3 (Continued)
3 结论
本研究利用磺胺类药物与荧光胺反应产生荧光基团的性质,建立了虾中14种磺胺类药物残留量的在线柱后衍生高效液相色谱分析方法。该法衍生反应效率较高,样品净化效果较好,待测物色谱图不存在杂峰干扰,灵敏度高,定性定量准确。14种磺胺类药物的定量限为1.0~5.0 μg/kg,能满足国内外对磺胺类药物的限量要求,适用于虾中痕量磺胺类药物的多残留检测。
[1] Zhang Y S,Nan H J,Wei X J.Animal Husbandry and Feed Science(张永生,南海娟,魏新军.畜牧与饲料科学),2009,30(3):35
[2] Wan C H,Long Z X.Modern Scientific Instruments(万春花,龙洲雄.现代科学仪器),2008,18(2):99
[3] Assil H I,Sheth H,Sporns P.Food Res Int,1992,25(5):343
[4] Zhang H Y,Cui X J,Geng J P,et al.Food Research and Development(张鸿雁,崔小军,耿金培,等.食品研究与开发),2008,29(9):181
[5] Tittlemier S A,Gelinas J M,Dufresne G,et al.Food Anal Methods,2008,1(1):28
[6] Casella I G,Contursi M,Gioia D.Electroanalysis,2012,24(11):2125
[7] Zotou A,Vasiliadou C.Chromatographia,2006,64(5):307
[8] Gehring T A,Griffin B,Williams R,et al.J Chromatogr B,2006,840(2):132
[9] Schneider M J,Braden S E,Reyes-Herrera I,et al.J Chromatogr B,2007,846(1/2):8
[10] Li C,Jiang H Y,Zhao S J,et al.Chromatographia,2008,68(1):117
[11] Sun L,Chen L G,Sun X,et al.Chemosphere,2009,77(10):1306
[12] Zhou M Y,Ma J,Gao X P,et al.Progress in Fishery Sciences(周明莹,马健,高湘萍,等.渔业科学进展),2011,32(2):102
[13] Granja R H M M,de Lima A C,Salerno A G,et al.Food Control,2012,28(2):304
[14] Liu H X.Food Science(刘海新.食品科学),2009,30(2):204
[15] Verzegnassi L,Savoy-Perroud M C,Stadler R H.J Chromatogr A,2002,977(1):77
[16] Li W H,Shi Y L,Gao L H,et al.Journal of Instrumental Analysis(厉文辉,史亚利,高立红,等.分析测试学报),2010,29(10):987
[17] Yu H,Tao Y F,Chen D M,et al.J Chromatogr B,2011,879(25):2653
[18] Wang L,Zhong D L,Chen G C,et al.Chinese Journal of Chromatography(王丽,钟冬莲,陈光才,等.色谱),2013,31(10):1010
[19] Zhang X J,Zheng B,Chen X C.Food Science(张小军,郑斌,陈雪昌.食品科学),2009,30(8):235
[20] Sílvia B,Ramon C,Jacinto G.J Agric Food Chem,2011,59(10):5240
[21] Lu K,Tong Q Y.Journal of Instrumental Analysis(卢坤,童群义.分析测试学报),2011,30(11):1320
[22] Zhang H Q,Song L L,Xu X L,et al.Chinese Journal of Analytical Chemistry(张海琪,宋琍琍,徐晓林,等.分析化学),2007,35(2):268
