过氧化氢异丙苯分解制苯酚的反应精馏工艺
2014-07-26周道伟叶建初林晗丹焦放健余光雄
周道伟,叶建初,林晗丹,焦放健,余光雄,沙 勇
(厦门大学化学化工学院,福建 厦门361005)
苯酚作为一种重要的化学品,每年的消耗量多达数百万吨.目前,90%以上的苯酚均通过异丙苯法制备,该工艺路线属于典型的反应+分离过程.异丙苯首先经氧化反应生成过氧化氢异丙苯(CHP),将氧化产物CHP和大量未反应的异丙苯混合物提纯浓缩后得到高浓度的CHP,再通过酸催化分解生成苯酚和丙酮[1-3].由于进入酸分解反应器的反应原料中还含有一定量的二甲基苄醇(DMBA),其在酸催化剂的作用下生成α-甲基苯乙烯(AMS)和水.分解反应器的出料进入后续的分离单元,对产物苯酚、丙酮进行分离,并回收AMS.
反应精馏作为一种工艺强化手段,具有高设备集成度和高反应热利用率等优势[4-7].由于CHP分解产物苯酚和丙酮的沸点相差大,因此可使用合适的固体酸催化剂,通过反应精馏技术实现CHP完全分解、利用反应热实现产物及时分离的双重目的.目前Exxon Mobli公司已报道了使用金属氧化物型超强酸作为催化剂的CHP分解反应精馏制苯酚技术[8].该过程使用提纯浓缩后的CHP作为原料,为移除CHP分解反应产生的大量反应热,向塔内引入大量丙酮作为热夹带剂.但是,由于丙酮的额外引入增加了分离负荷和设备投资,因此,尽管该CHP分解反应精馏技术具备技术上的先天优势,但其仍未在工业中广泛应用.
事实上,如果直接使用氧化得到的CHP和异丙苯混合物作为反应精馏塔进料,则可利用原料中含有的大量异丙苯作为热夹带剂,利用其汽化吸收热量移除反应热、控制塔内反应段温度,从而不必额外引入丙酮,可显著降低设备投资,并减少提纯浓缩CHP所需的能耗,进一步简化了生产工艺流程.此外,流程的改变使适用于该反应精馏工艺的催化剂种类得到扩展,即除了用于传统反应精馏工艺的金属氧化物型超强酸以及众多关于催化性能研究报道的沸石、蒙脱土及杂多酸等催化剂[9-11]外,阳离子交换树脂也将可作为该反应精馏工艺的催化剂.这主要是由于该工艺中未引入额外丙酮,因此反应段丙酮含量显著下降,从而使得阳离子交换树脂避免了因丙酮而出现溶胀、破碎的现象[12].
本文建立的苯酚生产反应精馏工艺将采用未经浓缩处理的CHP为原料,并选择阳离子交换树脂作为反应催化剂.通过对不同类型树脂催化剂的催化活性进行实验考察,将确定适合该工艺的树脂类型.在另一方面,将通过反应精馏小试实验来探究该反应精馏工艺在实际中的耦合可行性.
1 实验装置及方法
CHP、苯酚、丙酮、AMS以及异丁醇均购自国药集团化学试剂有限公司,异丙苯购自天津市光复精细化工研究所.以上原料中除作为反应原料的CHP外,其余均为分析纯.经碘量法分析,购置的化学纯CHP中CHP质量分数为84.5%.DMBA购自 Adamasbeta,纯度为98%,将作为色谱分析的标准品使用.
反应混合物的主要成分包含CHP、丙酮、异丙苯、苯酚、AMS和DMBA,利用以异丁醇为内标物的气相色谱法进行分析[11-12].色谱分析中采用的色谱柱为OV101毛细管柱(30m),气化温度和检测温度分别为210和250℃,柱炉起始温度为80℃,设定以10℃/min升至180℃,保持此温度2.5min.经气相色谱检测,原料CHP中含有8%(质量分数,下同)的DMBA.
选取陶氏化学公司的两种阳离子交换树脂作为催化剂,分别为湿型的AMBERLYSTTM35WET和干型的AMBERLYSTTM35DRY,形态均为直径0.8mm左右的球形固体.CHP催化分解动力学实验的反应装置如图1所示.图1中的蛇形冷凝管可将汽化产物冷凝回流至反应器.实验时将配比好的原料和树脂催化剂同时置于烧瓶中,使用恒温槽控制反应温度,利用机械搅拌以抑制外扩散对反应的不利影响.在综合考虑搅拌速率太快容易造成树脂的破碎后,本实验中设定搅拌速率为200r/min.随着反应的进行,通过定时从反应器中取样进行色谱分析,即可获知CHP催化反应的进程.
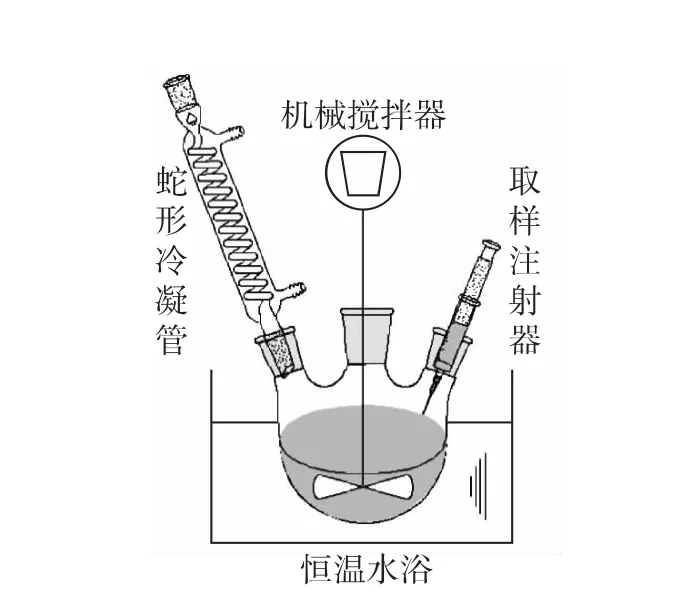
图1 CHP分解动力学实验装置Fig.1 Apparatus of kinetics experiment of CHP decomposition
为考察该反应精馏过程的实际可行性,建立了小试规模的连续反应精馏塔.该反应精馏塔为高度700 mm、塔径30mm的玻璃塔.该塔的上部为350mm的精馏段,下部为150mm的提馏段,两段均采用3mm×3mm的θ环进行填充.在提馏段以上设有200mm的反应段,进料口设置在反应段以上30mm处.反应段内除装填有θ环外,还混装一定量的树脂作为催化剂.为了固定树脂颗粒在塔内合适的位置,并避免树脂颗粒因碰撞造成破碎[13],需采用金属丝网对其进行封装处理.封装得到每个催化剂包内含树脂约0.15g,整个反应段共装有9个这样的催化剂包.
2 CHP催化分解的动力学实验
对于采用不同类型催化剂的动力学实验,反应物的初始组成、反应条件及催化剂用量列于表1.在4组实验中,反应温度均设定为65℃.实验中CHP和异丙苯组成比例接近于氧化得到的CHP和异丙苯混合物工业流股组成比例,苯酚在反应过程中随时间生成量如图2所示.

表1 CHP催化分解反应条件Tab.1 Experimental conditions of CHP catalytic decomposition reaction g

图2 不同实验条件下反应混合物中苯酚的含量Fig.2 Phenol content in reaction mixture under the different reaction condition
从图2可以看出,当体系初始丙酮质量分数较高且采用0.167g湿型树脂作为催化剂时,30min后体系内苯酚质量分数达到3.470%.但是,当初始丙酮质量分数为零时(实验2),即使采用1.959g湿型树脂作为催化剂,30min后体系内苯酚的质量分数也仅为0.600%.这意味着湿型树脂虽然适用于已报道的反应精馏工艺,但在本文提及的反应精馏工艺中明显不可用.之所以反应体系中丙酮质量分数具有如此显著影响,是由于CHP在酸分解过程中存在脱水的中间平衡步骤[1],而湿型树脂的含水量较高(可达50%),会对该步骤产生抑制作用,从而导致催化剂活性下降.
从图2还可发现,在湿型树脂预先进行脱水处理后(实验3),其催化活性有所上升,和实验1的结果相似.这进一步说明了,湿型树脂中所含水分对CHP酸催化分解反应的不利影响.
基于树脂中含水量对反应过程的影响,采用干型树脂进行了实验4.从图2可以看出,干型树脂由于具有非常低的含水量(小于3%),使其表现出最高的反应活性.在30min后,体系内苯酚的质量分数达到了5.063%,相对于使用湿型树脂作为催化剂的情况,使用干型树脂对产率具有明显的提高.而该类型树脂之所以可以表现出高于脱水后的湿型树脂,是由于湿型树脂的脱水过程存在相平衡的制约,无法实现水分的完全脱出.
由于干型树脂具有最优的催化活性并无需进行预脱水处理,因此在后续的反应精馏小试实验中,将采用干型阳离子交换树脂AMBERLYSTTM35DRY作为反应催化剂.
3 CHP制苯酚反应精馏实验
为了使精馏塔尽快达到CHP分解所需的反应条件,在进行CHP制苯酚反应精馏实验前将向塔釜一次性加入25℃,300mL不含CHP的混合液(苯酚13.62%,丙酮8.40%和异丙苯77.98%),并以100W的功率对塔釜加热,在全回流情况下操作45min.此后,精馏塔将按照表2所示的相关参数进行操作,并开始向精馏塔连续进料,具体进料组成如图3所示.

表2 反应精馏塔的实验条件Tab.2 Conditions of the reactive distillation experiment
需要特别说明的是,为了降低反应段的温度,抑制过程中的副反应并提高过程的安全性,反应精馏塔操作在57kPa的真空度下.在另一方面,由于在实际异丙苯氧化过程中副反应较多,这将导致氧化液中除主要产物CHP外,还含有一定量DMBA,其在树脂催化剂作用下,将分解为水和AMS[3],因此,为了让实验结果对实际工业过程更具参考价值,小试实验采用的进料为包含CHP、DMBA和异丙苯的混合物,其组成与氧化液组成相似[1-2,8].

图3 反应精馏塔及实验Fig.3 Reactive distillation column and experiment
CHP酸分解制苯酚反应精馏工艺的小试实验结果如图3所示.在设定的操作真空度下,塔顶和塔釜温度分别为43.3和126.9℃,较常压而言得到了显著降低.除真空度的作用外,体系内存在的大量异丙苯作为热夹带剂,其在塔顶和塔釜均有出现,吸收了大量反应热,一是整体上降低了塔内温度,二是塔顶和塔釜的分配比例将有效地调节精馏塔温度分布.此外,经气相色谱检测分析塔顶和塔釜组成中均不含CHP和DMBA,表明二者已完全反应,同时作为副产物的苯乙酮的质量分数也仅为1.00%,意味着此反应对产物苯酚的选择性较高.反应产物苯酚和丙酮可在塔内实现分离目的,在塔顶中苯酚质量分数仅为0.45%,丙酮在塔釜中质量分数仅为0.29%.实验结果表明该反应精馏过程可实现反应和分离的完美耦合.
4 结 论
选择阳离子交换树脂作为催化剂,研究了催化剂的类型和溶剂组成这两个因素对CHP酸催化分解过程的影响.本反应精馏工艺采用异丙苯的氧化液直接作为反应精馏塔的进料,而非经浓缩处理后的CHP.该工艺进料中异丙苯含量的上升导致了塔内反应段中丙酮含量显著降低.动力学实验结果表明,这样的工艺改变使得AMBERLYSTTM35DRY型树脂的催化活性明显高于AMBERLYSTTM35WET型树脂,即前者将更加适合于本工艺过程.本文为进一步研究该反应精馏工艺的实际可行性,建立了小试规模的连续操作反应精馏塔,其采用干型树脂作为催化剂,并在57 kPa的真空度下操作.小试实验结果表明,在反应精馏过程中,反应物CHP和DMBA的转化率均达到了100%,且可实现产物苯酚、丙酮的有效分离.
[1]Zakoshansky V.Method of producing phenol and acetone from cumene hydroperoxide:WO,0166500A1[P].2001-09-13.
[2]Levin D,Santiesteban J G,Vartuli J C.Production of phenol:US,6169215B1[P].2001-01-02.
[3]Schmidt R J.Industrial catalytic processes-phenol production[J].Applied Catalysis A:General,2005,280(1):89-103.
[4]An W Z,Meng Xia,Li H X,et al.Energy internal integration and optimization within ethoxylation reactive distillation column with consideration of heat of reaction[J].CIESC Journal,2012,63(11):3602-3608.
[5]Gao X,Li X G,Zhang R,et al.Process simulation of methyl acetate hydrolysis via catalytic distillation[J].CIESC Journal,2010,61(9):2442-2447.
[6]Zhao G S,Yang B L.Dehydration of tert-butyl alcohol in reactive distillation adapted to chemical heat pump[J].CIESC Journal,2004,55(3):384-389.
[7]Taylor R,Krishna R.Modelling reactive distillation[J].Chemical Engeering Science,2000,55(22):5183-5229.
[8]Doron L,Bala C,Jose G S.Production of phenol using reactive distillation:US,6410804B1[P].2002-06-25.
[9]Selvin R,Hus H L,Aneesh P,et al.Preparation of acidmodified bentonite for selective decomposition of cumene hydroperoxide into phenol and acetone[J].Reaction Kinetics Mechanisms Catalysis,2010,100(1):197-204.
[10]Selvin R,Rajarajeswari G R,Roselin L S,et al.Catalytic decomposition of cumene hydroperoxide into phenol and acetone[J].Applied Catalysis A:General,2001,219(1/2):125-129.
[11]Yadav G D,Asthana N S.Selective decomposition of cumene hydroperoxide into phenol and acetone by a novel cesium substituted heteropolyacid on clay[J].Applied Catalysis A:General,2003,244(2):341-357.
[12]Huang D G,Han M H,Wang J F,et al.Catalytic decomposition process of cumene hydroperoxide using sulfonic resins as catalyst[J].Chemical Engineering Journal,2002,88(1/2/3):215-223.
[13]Subawalla H,Gonzaléz J C,Seibert A F,et al.Capacity and efficiency of reative distillation bale packing:modeling and experimental validation[J].Industrial Engineering Chemical Research,1997,36(9):3821-3832.
