水羟硅钠石的制备与表征
2014-07-24戈明亮陈萌
戈明亮,陈萌
(华南理工大学聚合物成型加工工程教育部重点实验室,聚合物新型成型装备国家工程研究中心, 广东 广州 510640)
近年来,水羟硅钠石(Na2Si22O45·10H2O,kenyaite)及其相关科学的研究日益受到人们重 视[1-3]。Kenyaite 是一种二维层状结构硅酸盐材料,由Eugster 等[4]于1967 在肯尼亚magadi 湖中首次发现,它的层板是由带负电的[SiO4]四面体组成,层间有可被交换的水合钠离子[5],层板之间具有较好的膨胀性,可以容纳小到质子大到高分子和蛋白质等客体[6]。性能各异的客体分子同无机层状主体材料形成的复合材料在催化、吸附以及新型功能材料等领域具有重要的应用价值[7]。Alarcon 等[8]将Sn通过离子交换法插入到 kenyaite 层间,制备了Sn-kenyaite,该材料催化性能非常优异。Park 等[9]以介孔硅柱撑kenyaite 材料为催化剂载体,负载贵金属Ni、Pd 用于甲烷部分氧化,结果表明,甲烷具有较高的转化率且在750℃高温下加热100h 催化剂结构依然保持稳定。Guerra 等[10]将N-丙基乙烯三甲氧基硅烷和双-[3-(三乙氧基硅烷)丙基]-四硫化物分别插入到kenyaite 层间,制备了两种吸附材料,该吸附材料对有毒物质砷有较好的吸附效果,通过对比发现,[3-(2-氨乙基)氨丙基]三甲氧基硅烷改性的kenyaite 吸附砷的能力更强。
相比于蒙脱石等其他层状硅酸盐,kenyaite 结构中不含铝,具有更好的化学及热稳定性,离子交换能力更强。除此之外,与其制备过程中产生的中间相麦羟硅钠石(Na2Si14O29·9H2O,magadiite)相比其稳定性更好,层间距也更大,更利于客体分子的插层[11]。同时,通过常规的水热合成方法可以非常容易地在实验室中合成出kenyaite。目前,气相二氧化硅[12]、工业水玻璃[13]等均被用作硅源来制备kenyaite,但是这些硅源制备的kenyaite 存在成本过高或含有杂质等缺点,限制了kenyaite 的工业化应用。本实验以纯度高且价格低廉的沉淀白炭黑为硅源,采用水热法在SiO2-NaOH-Na2CO3-H2O 体系中制备了单一晶相kenyaite 并研究了其最佳的合成工艺,促进了kenyaite 的工业化应用。
1 实 验
1.1 样品制备
实验所用原料为分析纯的NaOH、Na2CO3和纯度为93%的沉淀白炭黑(7%H2O),实验用水为去离子水。按初始物料摩尔比为 n(SiO2) ∶n(NaOH+Na2CO3) ∶n(H2O)=7 ∶1 ∶100[ 固 定n(NaOH)∶n(Na2CO3)=1∶2]称取原料并混合搅拌15min,然后转移至聚四氟乙烯内衬的不锈钢反应釜中,170~180℃晶化反应9~48h,待反应釜冷却到室温后,将产物抽滤、水洗至pH=7~8,80℃下烘干4h 即得样品。其反应条件、初始物料比和实验结果列于表1。
1.2 样品表征
采用D8 ADVANCE 型X 射线衍射仪对样品进行物相分析[Cu 靶,管电压为40kV,管电流为40mA,扫描步长为0.02°,扫描速率为6°/min,扫描范围为3°~60°]。采用Nova Nano SEM 430型扫描电子显微镜对样品进行形貌分析(操作电压为10~20kV)。采用STA449 C 型同步热分析仪对样品进行热重-差热分析(升温速率为10℃/min,升温范围为室温~750℃)。使用NEXUS 670 型Fourier 变换红外光谱仪测定样品的红外光谱(波数范围为400~4000cm-1)。采用Axios PW4400 型X射线荧光仪对样品进行化学组分分析(功率为3.6kW,熔融制样,熔剂为Li2B4O7)。
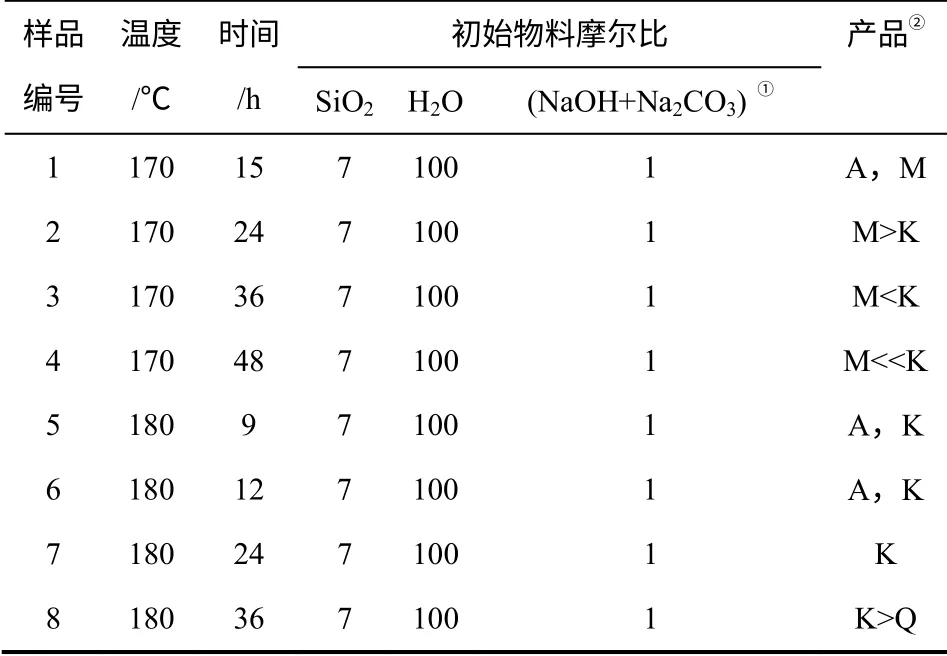
表1 反应条件和合成产物
2 结果与讨论
2.1 X 射线衍射分析
图1(a)和图1(b)分别是晶化温度为170℃ 和180℃时样品的X 射线衍射(XRD)谱。从图1(a)可以看出,在晶化时间为15h 时,样品中仅有magadiite 的特征峰;当晶化时间达到24h 时,样品中开始出现kenyaite 的特征峰,说明magadiite 开始向更稳定的kenyaite 转化;随着晶化时间进一步延长至36h,kenyaite 的特征峰进一步加强;但由于晶化温度较低,在晶化时间达到48h 时,样品中仍存在magadiite 的特征峰。由图1(b) 可以看出,在晶化时间为9h 时,出现较弱特征峰,此峰归属于kenyaite 峰,说明当晶化温度足够高时,kenyaite 可以不经历中间相magadiite 而直接生成;当晶化时间延长至12h 时,kenyaite 的特征峰有所加强,但样品晶化尚未完成;当晶化时间达到24h 时,kenyaite晶化已经完成,将该XRD 谱与已知的kenyaite 的XRD 谱进行对比[14],整个谱图与kenyaite 的特征衍射峰相吻合,2θ=4.545°,层间距为1.94 nm,确定该晶相为单一晶相kenyaite;当晶化时间达到36h时晶化,产物中出现了石英相,这说明kenyaite 开始向更稳定的石英转化。结合图1(a)和图1(b)可以看出,当反应温度由170℃升高到180℃时,尽管反应温度只增加了10℃,但反应产物却发生了剧烈变化,说明温度是影响kenyaite 合成的主要因素[15]。综上所述,kenyaite 的最佳晶化合成条件为晶化温度为180℃,晶化时间为24h。

图1 不同晶化温度和晶化时间下制备的样品XRD 谱
2.2 显微结构分析
图2 是最佳晶化合成条件下制备的kenyaite 的扫描电镜(SEM)照片,由图2 可知,kenyaite 的形状为玫瑰花瓣状,这与之前的报道一致[16]。
2.3 红外光谱分析

图2 kenyaite 的扫描电镜照片
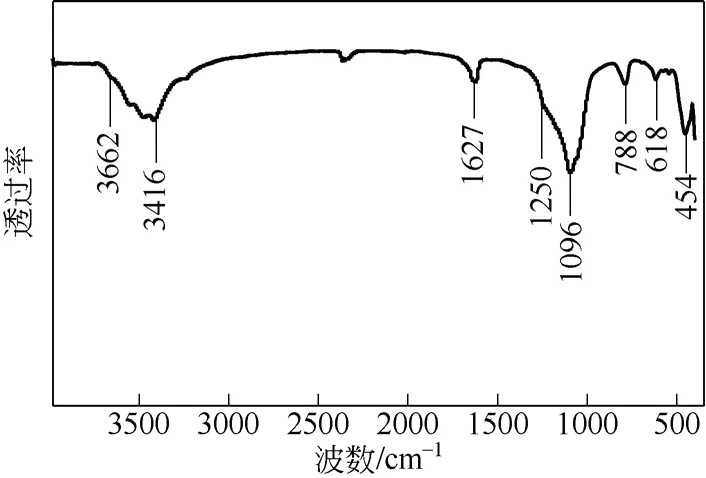
图3 kenyaite 的红外光谱
图3 为最佳晶化合成条件下制备的kenyaite 的红外(IR)光谱。其中,3662cm-1和3418cm-1处的 吸收峰对应缔合O—H 键的伸缩振动,1627cm-1处的吸收峰对应水的弯曲振动,1250cm-1处的吸收峰为Si—O—Si 的反对称伸缩振动吸收峰,该峰表明kenyaite 结构中存在五元环[15],1096cm-1和788cm-1处的吸收峰则为Si—O 四面体的对称和反对称伸缩振动峰,618cm-1处的吸收峰是由于双环的振动,而在454cm-1处的吸收峰证明了kenyaite 中包含着Si—O 六元环[17]。
2.4 同步热分析
图4 为最佳晶化合成条件下制备的kenyaite 的同步热分析(STA,TG-DSC 联用)曲线。从图4可见,kenyaite 失重分为两步。第一步为30~350℃,是一个吸热过程,失重为10.12%,为脱除吸附水阶段。第二步在350℃以上,是一个吸热过程,失重为0.41%,这部分的失重可能是由羟基的缩合反应造成的,这也可能是kenyaite 结构发生改变的原因[15]。综上所述,样品的热稳定性在350℃以下。
2.5 化学组分分析
采用X 射线荧光(XRF)光谱仪对最佳晶化合成条件下制备的kenyaite 进行化学组分分析,分析结果如表2 所示,所合成的kenyaite 的化学组成为Na2.03Si22O45.02·8.89H2O(所含吸附水的量随处理方
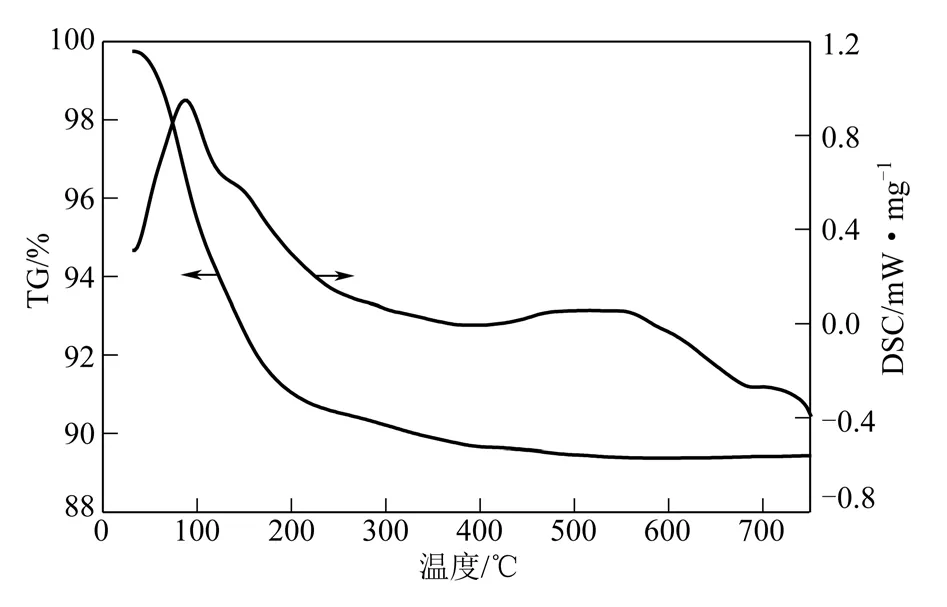
图4 kenyaite 的STA 图

表2 kenyaite 的主要化学组成
3 结 论
(1)采用水热法在SiO2-NaOH-Na2CO3-H2O体系下合成了单一晶相kenyaite。
(2)温度是影响kenyaite 制备的主要因素。
(3)kenyaite 的最佳晶化合成条件为晶化温度为180℃,晶化时间为24h。
(4)SEM 照片和TG-DSC 表明kenyaite 的形状为玫瑰花瓣状且其热稳定性在300℃以下。
[1] Royer B,Cardoso N F,Lima E C. et al. Organofunctionalized kenyaite for dye removal from aqueous solution[J]. J. Colloid Interface Sci.,2009,336 :398-405.
[2] Ruiz V S.O,Petrucelli G C,Airoldi C. Inorganic-organic hybrids derived from lamellar acidic kenyaite immobilizations for cation removal at the solid/liquid interface[J]. J. Mater. Chem.,2006,16:2338-2346.
[3] Kwon O Y,Park K W. The preparation of flaky layered carbon by using layered silicate template[J]. B. Kor. Chem. Soc.,2003,24(11):1561-1562.
[4] Eugster H P. Hydrous sodium silicate from Lake Magadi,Kenya,precursors of bedded chert[J]. Science,1967,157:1177-1179.
[5] Almond G G,Harris R K,Franklin K R. A structural consideration of kanemite,octosilicate,magadiite and kenyaite[J]. J. Mater. Chem.,1997,7(4):681-687.
[6] Wang Z,Pinnavaia T J. Intercalation of poly(propyleneoxide) amines (Jeffamines) in synthetic layered silicas derived from ilerite,magadiite,and kenyaite[J]. J. Mater. Chem.,2003,13:2127-2131.
[7] Price P M,Clark J H,Macquarrie D J. Modified silicas for clean technology[J]. J. Chem. Soc.,Dalton Trans.,2000,101-110.
[8] Villa de P A L,Alarcon C,Montes de C C. Nopol synthesis over Sn-MCM-41 and Sn-kenyaite catalysts[J]. Catal. Today,2005,107-108:942-948.
[9] Park K W,Jung J H,Seo H J,et al.Mesoporous silica-pillared kenyaite and magadiite as catalytic support for partial oxidation of methane[J]. Microporous Mesoporous Mater.,2009,121:219-225.
[10] Guerra D L,Airoldi C,Viana R R. Adsorption of arsenic(V) into modified lamellar Kenyaite[J]. J.Hazard Mater.,2009,163:1391-1396.
[11] 戈明亮,陈萌. 麦羟硅钠石的制备与表征[J]. 硅酸盐学报,2013,41(12):1704-1708.
[12] 刘子玉,刘中民,等. 凝胶硅铝比对合成 MCM-22,UTM-1 和Kenyaite 的影响[J]. 催化学报,2009,38(1):120-123.
[13] Kwon O Y,Park K W. Synthesis of layered silicate from sodium silicate solution[J]. B. Kor. Chem. Soc.,2004,25(1):25-26.
[14] Feng F X,Kenneth J,Balkus J R. Synthesis of kenyaite,magadiite and octosilicate using poly(ethylene glycol) as a template[J]. J. Porous Mater.,2003,10:5-15.
[15] Kwon O Y,Jeong S Y,Suh J K,et al. Hydrothermal syntheses of Na-magadiite and Na-kenyaite in the presence of carbonate[J]. B. Kor. Chem. Soc.,1995,16(8):737-741.
[16] Kwon O Y,Choi S W. Silica-pillared H-kenyaites:Interlamellar base catalyzed-reaction of tetraethylorthosilicate in water suspension[J]. B. Kor. Chem. Soc.,1999,20(1):69-75.
[17] Huang Y N,Jiang Z M,Schwieger W. Vibrational spectroscopic studies of layered silicates[J]. Chem. Mater.,1999,11:1210-1217.
