常染色体显性遗传眼球震颤家系的临床表型及致病基因的研究△
2014-07-24布娟刘敬李爱军陆遥庞宏蕾刘峰王乐今
布娟 刘敬 李爱军 陆遥 庞宏蕾 刘峰 王乐今
【应用研究】
常染色体显性遗传眼球震颤家系的临床表型及致病基因的研究△
布娟 刘敬 李爱军 陆遥 庞宏蕾 刘峰 王乐今
注:布娟、刘敬同为第一作者!
About BU Juan:Female,born in March,1973.Medical doctor.Tel:13717531820;E-mail:bujuan110@163.com
About LIU Jing:Female,born in May,1971.Medical doctor.Tel:13811854669;E-mail:jingliuliqun@sina.com
Accepted date:Jul 29,2014Foundation item:National Natural Science Foundation of China(No:81300789)From theDepartmentofOphthalmology,theThirdHospitalofPekingUniversity,Beijing100191,China
Responsible author:WANG Le-Jin,E-mail:wanglejin_puec@sina.com
先天性眼球震颤;常染色体显性遗传;PAX6基因;FRMD7基因;GPR143基因
目的 研究1个中国人常染色体显性遗传性眼球震颤家系的临床特点,并通过候选基因直接测序的方法对该家系的致病基因及发病机制进行研究。方法 选择1个先天性眼球震颤家系,对家系所有成员进行全身检查及视力、眼位、眼球运动、眼球震颤中间带、验光等眼科的相关检查后,从家系中每一代各选1例患者(包括先证者)及正常人,进行候选基因FRMD7、GPR143与PAX6基因的外显子测序。结果 家系患者的眼球震颤为水平冲动型,并且具有中间带。除了先证者有部分眼组织缺损及小眼球等异常外,其余患者的眼前节均未见明显发育异常。家系患者PAX6基因第7外显子的第382碱基发生了杂合突变(c.C382T),从而引起了氨基酸的改变(p.R128C),该突变可能影响了PAX6蛋白与其调控的下游基因的调控序列的结合,进而导致PAX6基因功能异常,影响眼部发育。结论 该常染色体显性遗传性眼球震颤家系的致病基因为PAX6基因。
[眼科新进展,2014,34(11):1042-1044]
先天性眼球震颤(congenital nystagmus,CN)常发生于刚出生时或生后3个月内,表现为不自主地、节律性或往复性地眼球摆动或跳动。在CN患者中,有少部分人在向某一方向注视时,眼球震颤的幅度及频率明显减轻,即存在中间带,称为先天性特发性眼球震颤(congenital idiopathic nystagmus,CIN)。有7% ~30% 的CIN患者具有遗传倾向,最常见的遗传方式是X染色体连锁遗传。近年来,随着分子遗传学技术迅速发展,有关CIN的基因定位与致病基因克隆取得了很大的进展[1-2]。已知的致病基因有酵母功能域包含蛋白7(FRMD7)和GPR143。
本研究收集了1个4代CIN家系,其遗传方式是较为少见的染色体显性遗传,对家系患者进行了临床表型的研究和FRMD7、GPR143、PAX6基因的突变筛查。现将研究结果报告如下。
1 资料与方法
1.1 一般资料 研究对象取自在北京大学眼科中心就诊的一个CIN大家系,家系68名成员中共有11例患者,其遗传方式符合常染色体显性遗传。本研究遵循赫尔辛基宣言,在家系所有成员均知情同意的情况下,抽取家系中11例患者和12例正常人的外周血8~10 mL,并对他们进行全面的检查。
1.2 方法
1.2.1 眼科检查 详细询问病史及家族史后进行视力、眼位、眼球运动、眼球震颤中间带、验光等相关检查。使用复方托吡卡胺滴眼液散瞳进行裂隙灯显微镜和检眼镜检查以排除眼前节及眼底病变。根据临床体征和检查结果,确定患者和正常人。诊断标准:第一眼位出现显性眼球震颤者为阳性。
1.2.2 基因组DNA的提取 抽取外周血8~10 mL,分离提取基因组DNA,紫外分光光度计和6 g·L-1琼脂糖凝胶电泳检测样本DNA纯度和浓度。加入1000 μL TE,-20℃保存备用。
1.2.3 FRMD7、GPR143与PAX6基因突变检测 从家系中每一代各选1例患者及正常人,其中一人为先证者,通过下列引物对FRMD7、GPR143与PAX6基因的外显子(exon)、5’和3’ 端非编码区(untranslated region,UTR)以及内含子(intron)与外显子拼接处序列进行PCR反应扩增。PCR反应总体系为:10×缓冲液5 μL(Mg2+浓度为1.5 mmol·L-1),4×dNTP 8 μL(2.0 mmol·L-1),上、下游引物各0.5 μL(20 pmol·μL-1),Taq DNA聚合酶0.5 μL(5 U·μL-1),模板DNA 1 μL(100 ng·μL-1),加消毒双蒸水至50 μL。PCR反应条件:94 ℃预变性5 min,94 ℃变性 30 s,60~62 ℃退火30 s,72 ℃延伸45 s后延伸7 min,30~35个循环。PCR反应结束后,取5 μL PCR扩增产物进行琼脂糖凝胶电泳以观察扩增效率,剩余PCR扩增产物经20 g·L-1琼脂糖凝胶电泳进行纯化回收后直接进行测序。
2 结果
2.1 眼科检查 经家系系谱调查和全面的眼科检查发现,该家系中发病者在4代系谱中连续出现,患者双亲中有一方患病,则患者子女中有1/2发病,且男女患病机会均等,符合常染色体显性遗传特点。该家系发病的特点是出生后即双眼患病(家系患者的临床表型见表1),双眼水平冲动型眼球震颤,具有中间带及代偿头位。双眼在头正位下的矫正视力为0.10~0.60,双眼在代偿头位下的矫正视力为0.25~1.00。裂隙灯显微镜检查仅先证者有部分虹膜缺损、脉络膜缺损、小眼球,其余患者眼前节均未见明显发育异常。所有患者均未合并全身其他系统异常。
表1 家系患者眼科检查
Table1 Ophthalmologic test of both eyes of affected individuals with nystagmus

CodeSexAge/yearCorrectedvisualacuity(OU)NormalpositionAbnormalheadpositionDiagnosisⅢ-1Female780.300.60Nystagmus、Cataract(OU)Ⅲ-7Female640.100.25Nystagmus、Cataract(OU)Ⅲ-12Male600.200.40Nystagmus、Cataract(OD)Ⅳ-1Female550.600.80NystagmusⅣ-3Female530.300.60NystagmusⅣ-5Female470.601.00NystagmusⅣ-16(proband)Female380.250.30Nystagmus、Congenitalmicrophthalmos、Cataract(OU)Colobomaofirisandchoroid(OU)Ⅳ-18Male350.500.80NystagmusⅣ-22Male410.400.60NystagmusⅤ-1Female310.600.80NystagmusⅤ-4Male290.400.80Nystagmus
2.2 基因测序结果
2.2.1 FRMD7基因、GPR143基因测序 家系患者FRMD7基因、GPR143基因所有外显子以及内含子与外显子拼接处序列测序均未见突变。
2.2.2 PAX6基因测序 先证者及家系患者PAX6基因第7外显子的第382碱基发生了杂合突变(c.C382T),从而引起了氨基酸的改变(p.R128C,图1)。该突变在家系正常人及正常对照者中未检出。
3 讨论
由于CN发生于视觉发育早期,故而对患者的双眼视功能造成严重的损害。部分CN患者由于存在静止眼位或中间带(即当双眼向某一方向注视时眼球震颤可明显减轻或消失),眼部也无异常改变,患者常采取代偿头位。随着分子生物学的发展,对于这部分眼球震颤病例的发病机制研究有了很大的进展。
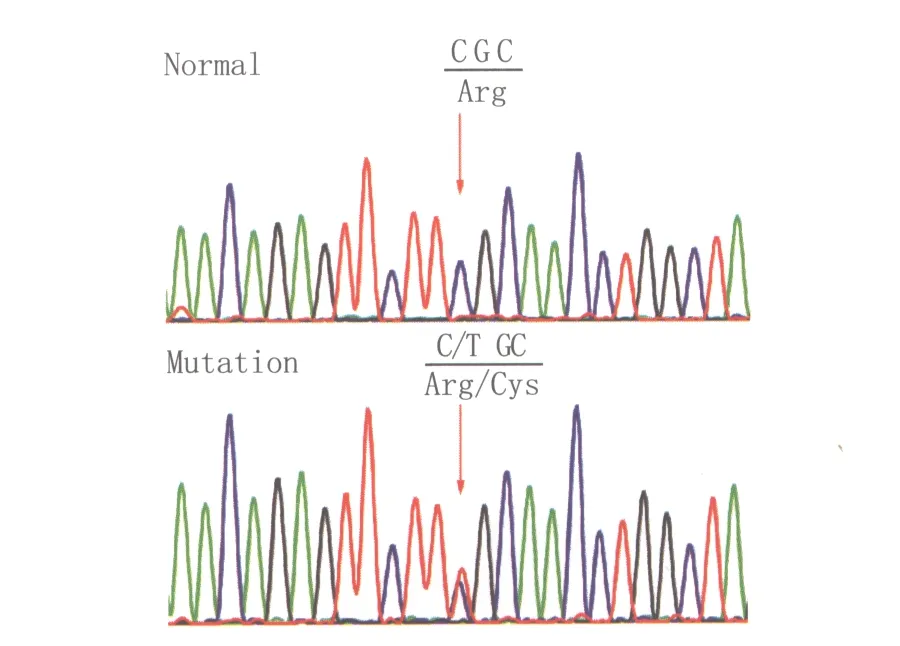
Figure 1 The PAX6 sequence of the patients with nystagmus 眼球震颤患者PAX6 基因测序结果
CIN的遗传方式以X染色体连锁遗传最常见。由于CIN具有很高的遗传异质性,且遗传的复杂性、染色体的随机失活使其分子机制的研究更为复杂。目前,已知的与CIN有关的致病基因有FRMD7和GPR143基因。
FRMD7基因有12个外显子,编码714个氨基酸,组成FRMD7,是超家族蛋白的成员之一。FRMD7基因主要在早期胚胎(孕56 d)的前脑室、中脑、小脑原基、脊索及正在发育的神经视网膜中表达,在孕37 d胚胎则主要在中脑和后脑表达,而此区域被视为眼球运动控制中枢所在地[3-5]。
GPR143基因包含9个外显子,编码404个氨基酸,含有7个跨膜区域,属于G蛋白耦联受体。GPR143基因最初被鉴定为眼白化病致病基因,其突变会引起眼球色素的减少、畏光和视敏度的降低。GPR143基因主要表达在皮肤和眼的色素细胞上,它位于细胞内黑色素小体膜上,调节黑色素的发生和成熟[6-7]。关于GPR143基因突变导致CIN 的确切发病机制尚不清楚,推断GPR143基因突变可能阻断了一条重要的信号转导通路,造成眼球运动传导系统功能缺陷[8-9]。
PAX6基因编码一个含有422个氨基酸的转录因子,该转录因子属于同源异型框蛋白家族成员,主要调控眼睛的发育和其他发育过程。PAX6基因定位于染色体11q13~14区间,包含13个外显子。PAX6蛋白包含配对结构域、同源异型结构域、富含脯氨酸-丝氨酸-苏氨酸的结构域[10]。PAX6 基因是眼球发育中极为重要的基因,是绝大多数先天性无虹膜病例的致病基因[11-16],在 PAX6 杂合突变眼中,眼前段组织发育受到严重的损害,如小梁网分化不良、角膜上皮层变薄、虹膜异常、白内障等。
本研究鉴定的p.R128C突变位于配对结构域的C末端,突变可能影响了PAX6蛋白与其调控的下游基因的调控序列的结合,进而引起与眼睛发育相关基因的表达异常,从而导致该家系先证者的眼球震颤、白内障、小眼球、虹膜缺损等临床表现。
PAX6基因的p.R128C突变曾被报道过2次,Azuma等[17]在一个日本的常染色体显性遗传的单纯性中心凹发育不良家系中鉴定了一个p.R128C突变,该家系的所有患者均表现为先天性的单纯性中心凹发育不良伴随正常的眼前段结构以及眼球震颤,中心凹光反射消失,眼底图像显示视网膜血管穿过中心凹区域。van Heyningen等[18]在一个欧洲的病例中鉴定了同样的突变,患者具有和Azuma等[17]报道的一样的表型。但在我们的研究中,家系内患者表现出不同于以往报道的临床表型,所有患者均表现为CIN,除了先证者还表现出白内障、小眼球、虹膜缺损等眼前段结构异常外,其余患者并无眼前节及眼底的异常。这提示同样的突变在不同的遗传环境下可能会引起不同的临床表型,所以本研究所鉴定的PAX6蛋白p.R128C突变进一步证实了PAX6基因突变的临床异质性。
由于CN目前尚无有效的治疗方法,只有对该病的不同遗传方式和发病机理有了清晰认识,才能够利用遗传学手段为CN患者提供准确的遗传咨询和产前诊断服务,探索新的治疗途径。
1 Self J,Lotery A.A review of the molecular genetics of congenital Idiopathic Nystagmus(CIN)[J].OphthalmicGenet,2007,28(4):187-191.
2 Gottlob I,Proudlock FA.Aetiology of infantile nystagmus[J].CurrOpinNeurol,2014,27(1):83-91.
3 Zhang X,Ge X,Yu Y,Zhang Y,Wu Y,Luan Y,etal.Identification of three novel mutations in the FRMD7 gene for X-linked idiopathic congenital nystagmus[J].SciRep,2014,17(4):3745-3748.
4 Pu J,Mao Y,Lei X,Yan Y,Lu X,Tian J,etal.FERM domain containing protein 7 interacts with the Rho GDP dissociation inhibitor and specifically activates Rac1 signaling[J].PLoSOne,2013,8(8):e73108.
5 Watkins RJ,Thomas MG,Talbot CJ,Gottlob I,Shackleton S.The Role of FRMD7 in Idiopathic Infantile Nystagmus[J].JOphthalmol,2012,2012:460956.
6 Camand O,Boutboul S,Arbogast L,Roche O,Sternberg C,Sutherland J,etal.Mutational analysis of the OA1 gene in ocular albinism[J].OphthalmicGenet,2003,24(3):167-173.
7 Micale L,Augello B,Fusco C,Turturo MG,Granatiero M,Piemontese MR,etal.GPR143 mutational analysis in two Italian families with X-linked ocular albinism[J].GenetTestMolBiomarkers,2009,13(4):527-531.
8 Peng Y,Meng Y,Wang Z,Qin M,Li X,Dian Y,etal.A novel GPR143 duplication mutation in a Chinese family with X-linked congenital nystagmus[J].MolVis,2009,15:810-814.
9 Hu J,Liang D,Xue J,Liu J,Wu L.A novel GPR143 splicing mutation in a Chinese family with X-linked congenital nystagmus[J].MolVis,2011,17:715-722.
10Glaser T,Ton CC,Mueller R,Petzl-Erler ML,Oliver C,Nevin NC,etal.Absence of PAX6 gene mutations in Gillespie syndrome(partial aniridia,cerebellar ataxia,and mental retardation)[J].Genomics,1994,19(1):145-148.
11Robinson DO,Howarth RJ,Williamson KA,van Heyningen V,Beal SJ,Crolla JA.Genetic analysis of chromosome 11p13 and the PAX6 gene in series of 125 cases referred with aniridia[J].AmJMedGenetA,2008,146(5):558-569.
12Thaung C,West K,Clark BJ,McKie L,Morgan JE,Arnold K,etal.Novel ENU-induced eye mutations in the mouse:models for human eye disease[J].HumMolGenet,2002,11(7):755-767.
13Tang SM,Rong SS,Young AL,Tam PO,Pang CP,Chen LJ.PAX6 gene associated with high myopia:a meta-analysis[J].OptomVisSci,2014,91(4):419-429.
14Lee HJ,Colby KA.A review of the clinical and genetic aspects of aniridia[J].SeminOphthalmol,2013,28(5-6):306-312.
15Thomas S,Thomas MG,Andrews C,Chan WM,Proudlock FA,McLean RJ,etal.Autosomal-dominant nystagmus,foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation[J].EurJHumGenet,2014,22(3):344-349.
16Peter NM,Leyland M,Mudhar HS,Lowndes J,Owen KR,Stewart H.PAX6 mutation in association with ptosis,cataract,iris hypoplasia,corneal opacification and diabetes:a new variant of familial aniridia[J]?ClinExperimentOphthalmol,2013,41(9):835-841.
17Azuma N,Nishina S,Yanagisawa H,Okuyama T,Yamada M.PAX6 missense mutation in isolated foveal hypoplasia[J].NatGenet,1996,13(2):141-142.
18van Heyningen V,Williamson KA.PAX6 in sensory development[J].HumMolGenet,2002,11(10):1161-1167.
date:May 26,2014
Clinical and genetic study of autosomal dominant congenital nystagmus family
BU Juan,LIU Jing,LI Ai-Jun,LU Yao,PANG Hong-Lei,LIU Feng,WANG Le-Jin
congenital nystagmus;autosomal dominant;PAX6 gene;FRMD7 gene;GPR143 gene
Objective To study the clinical phenotype in a Chinese family with congenital nystagmus and determine the disease-causing mutation.Methods A detailed clinical ophthalmic and complete physical examinations were performed for all patients in the congenital nystagmus family.Mutations in PAX6,FRMD7 and GPR143 were determined by PCR-based DNA sequencing assays and multiplex PCR assays for deletions.Results The affected members in the pedigree had classical phenotype of nystagmus.The type of nystagmus in the family was jerk nystagmus with null-point.Only the proband had ocular coloboma and congenital cataract.The missense mutation R128C(caused by a 382 C→T heterozygous change) in PAX6 was identified in the Chinese congenital nystagmus family.The mutation was associated with the disease phenotype in patients,but not detected in their relatives or in the 100 normal controls.Conclusion The missense mutation of PAX6 is the molecular basis for the congenital nystagmus family.
布娟,刘敬,李爱军,陆遥,庞宏蕾,刘峰,等.常染色体显性遗传眼球震颤家系的临床表型及致病基因的研究[J].眼科新进展,2014,34(11):1042-1044.
10.13389/j.cnki.rao.2014.0288
布娟,女,1973年3月出生,山东人,医学博士,副主任医师。主要研究方向为斜视与小儿眼科、眼科遗传学。联系电话:13717531820;E-mail:bujuan110@163.com
作者简介:刘敬,女,1971年5月出生,山东人,医学博士,主治医师。研究方向为斜视与小儿眼科学、眼科遗传学。联系电话:13811854669;E-mail:jingliuliqun@sina.com
2014-05-26
国家自然科学基金资助(编号:81300789)
100191 北京市,北京大学第三医院眼科
王乐今,E-mail:wanglejin_puec@sina.com
修回日期:2014-07-29
本文编辑:付中静
[Rec Adv Ophthalmol,2014,34(11):1042-1044]
