手性化合物酶法制备中辅酶再生体系的构建与应用进展
2014-07-18彭益强张曙伟
彭益强,张曙伟
(华侨大学工业生物技术福建省高等学校重点实验室,福建 厦门 361021)
手性化合物是有机合成中不可缺少的重要添加剂,也是制备具有光学活性的医药、农药和功能材料的重要中间体,在生理生化、化学合成中具有重要意义。如手性醇是一类重要的手性模块化合物,在医药、农业、材料、有机中间体以及环保等领域都具有广泛的应用价值[1-5]。
近年来,生物法合成手性化合物已成为制备手性中间体的研究热点,如许多手性醇可以通过氧化还原酶催化潜手性酮的立体选择性还原得到[6-10]。但是,利用酶催化不对称还原反应的一个瓶颈是需要解决反应体系中辅酶供应问题[11-12]。氧化还原酶的辅酶大多为烟酰胺类,主要是 NAD+/NADH 和NADP+/NADPH。在工业生物催化应用中,通常需要人为添加外源性辅酶以实现氧化还原酶的连续多批次催化,而烟酰胺类辅酶价格昂贵、稳定性差、难以重复利用,导致反应成本高昂。
为了解决手性化合物酶法还原制备反应中辅酶需求问题,学者们提出了各种辅酶再生的方法,如酶法再生、电化学法再生和光化学法再生等[13-15],其中具有较大工业应用潜力的是酶法再生,而酶法再生又可根据辅酶再生体系形成的环境分为胞内酶催化再生与胞外酶催化再生。本文主要介绍酶法再生中辅酶再生体系构建方式进展与应用实例,评价各种辅酶再生体系在工业应用中的特点,提出辅酶再生体系应用中可能遇到的问题与解决方法,希冀对手性化合物的酶法催化制备提供良好的解决方案。
1 单细胞内辅酶再生系统
生物细胞是酶制备与酶催化的最基本体系,细胞体内的酶代谢反应存在着一定协同性与可塑性,因此可在细胞内利用天然或改造的代谢途径中的相关酶实现辅酶在细胞体内的自我再生。
1.1 利用内源性辅酶再生系统
在细胞的生长代谢中,为了维持还原代谢途径的还原力的供给,有些细胞本身存在提供还原力的耦合代谢途径,从而在持续转化还原酮底物过程中,可从细胞内另一氧化途径中连续获得还原力质子,从而保证了手性化合物的连续发酵制备。
如 Dirk Weuster-Botz[16]利用蓝细菌聚球藻(SynechococcusPCC7942)中的脱氢氧化酶还原五氟苯乙酮(2'-3'-4'-5'-6'-pentafluoroacetophenone,PFAP)制备S型的五氟苯乙醇[pentafluoro (phenly-)ethanol,S-PFE],在无光条件下蓝细菌聚球藻可利用胞内的葡萄糖氧化脱氢反应途径为S-PFE的制备反应提供还原力质子,而同时利用胞内的葡萄糖代谢途径很容易实现NADPH的再生,只需外加葡萄糖和ATP,克服了光照条件的限制,可以持续获得ee值>99%的立体化合物。又如Gao等[17]应用氧化葡萄酸杆菌(Gluconobacter oxydans)经两步(先脱氢成醛再加羟基成羧酸)转化DL型丙二醇(DL-1,2-propanediol)制备 D 型 2-羟基丙酸(2-hydroxypropanic acid),同时利用频哪酮(3,3-二甲基-2-丁酮)的还原反应实现辅酶NAD(P)+的再生(图 1)。游离细胞通过离心可反复发酵利用,转化24h后反应达到平衡,D型2-羟基丙酸的获得率提高了36%,ee值达99%。
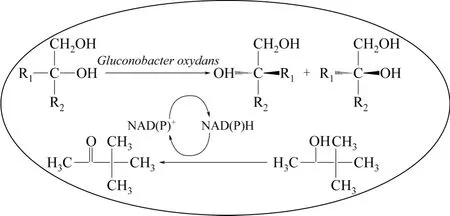
图1 葡萄杆菌(Gluconobacter oxydans)中利用胞内酶催化实现辅酶NAD(P)+再生
利用细胞自身代谢反应实现酶催化的耦合进行辅酶再生,这种方法简便易行,操作简单且可以实现多批次发酵转化反应。存在的问题是菌株的选择面较窄,虽然大多数菌株存有辅酶代谢的反应路径,但其本身不一定是为了满足人为设置的催化途径所需的氧化还原力要求。另外,这种利用细胞内部代谢路径进行催化反应与辅酶再生的方法,由于细胞中存在其他众多代谢途径,因此往往代谢副产物较多,目的产物的手性纯度不高。
1.2 利用代谢工程技术
微生物细胞中辅酶的生成与降解是处于一个动态的平衡过程,因此可以对微生物细胞内本身存在的 NAD(P)合成途径和补救途径加以改造,通过直接调控内源性辅酶量的方法来解决手性化合物制备过程中辅酶供应问题[13]。如Liang等[18]在野生型的大肠杆菌(E.coliW1485)中过量表达尼克酸磷酸核糖酰基转移酶(nicotinic acid phosphoribosyltransferase,NAPRTase)构建代谢工程菌E.coliNZN111,可调控 NADH池的大小与NADH/NAD+的比例从而提高代谢终产物琥珀酸的产量,该课题组同时也在E.coli中的NADH合成路径中过量表达尼克酸磷酸核糖酰基转移酶(NAPRTase)同时敲除ldhA和pflB基因片段,也可降低了 NAD(H)池中 NADH/NAD+的比例,一样提高了还原代谢途径终端代谢产物琥珀酸的产量[19]。
上述方法是直接调控辅酶的量,有时针对性并不是很强。Schroer等[20]通过在巴斯德毕赤酵母(Pichia pastoris)的甲醇代谢途径中过量表达甲醛脱氢酶(Formaldehyde dehydrogenase,FLD),既抑制了甲醛向二羟基丙酮方向代谢又克服了整个代谢途径中辅酶再生的瓶颈,加速了NADH的再生速度,而此代谢途径与3-羟基丁醇(acetoin)加氢还原生成2,3-丁二醇(2,3-butanediol)的反应相耦合,为 2,3-丁二醇的生产制备提供了充足的还原力(图2)。

图2 巴斯德毕赤酵母(Pichia pastoris)中通过代谢调控加速辅酶NAD+再生
1.3 利用基因工程技术
利用基因重组技术直接在宿主细胞中表达主催化酶与辅酶再生酶,从而构建同时具有催化转化功能与辅酶再生能力的工程细胞。这种方法针对性极强,融合了理论设计的酶耦合催化优势和细胞发酵催化转化的优势,使微生物的改造从随机突变发展到定向设计阶段。实践当中也有许多构建成功的例子[21-24]。但重组菌要应用于工业生产还存在着诸如底物浓度较低(底物在菌液中溶解度有限,高浓度底物对重组菌的毒性)、辅助底物及昂贵诱导物IPTG的添加问题等几个困难[25]。
为了克服有机底物在水相中溶解度较低问题,可以将能有效溶解有机底物的较温和的溶剂,如水溶性离子液体与细胞所处的水相反应体系相结合构建双相反应体系。这种方法既保证了细胞催化酶的活性同时又加大了底物的溶解度,提高了基因工程细胞的转化能效果。Bratigama 等[26]从12种离子液体中经筛选得到最适合底物溶解的离子液体[HMPL][NTF],同时将共表达来源于短乳杆菌(Lactobacillus brevis)的醇脱氢酶(alcohol dehydrogenase,LB-ADH)和源于博伊丁假丝酵母(Candida boidinii)的甲酸脱氢酶(formate dehydrogenas,CB-FDH)的基因工程菌[E.coliBL21(DE3)]在水相中进行催化转化各辅酶再生,催化辛酮合成辛醇,既可以克服底物辛酮低溶解度问题,又可以利用不同相的差别构建了底物储存和产物原位提取的集成过程(图 3)。在进行了一个批次的转化反应后,菌株转化的空时收率达 180g/(L·d),化学收率为95%,ee值达99.7%。
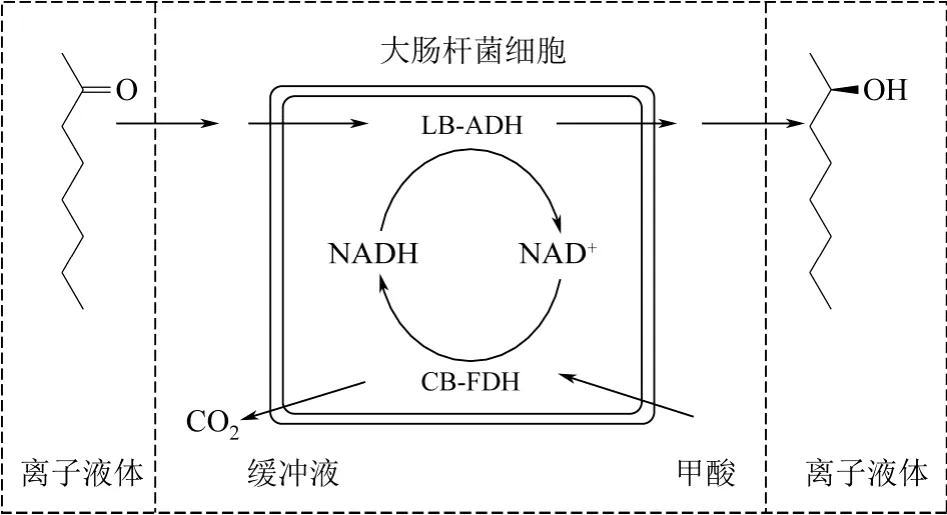
图3 构建有辅酶再生体系的基因工程菌E.coli 在离子液体-缓冲液双相体系中的催化
另外,为了解决高浓度底物的问题,也可以在生产工艺过程中采取底物批次添加策略。Ni等[27]在E.coli中共表达源于芽孢杆菌属(Bacillussp.)的羰基还原酶(carbonyl reductase,CR)和葡萄糖脱氢酶(glucose dehydrogenase,GDH),构建了双酶共表达体系,应用此工程菌转化底物乙烷-4-氯-3-氧桥丁酸(ethyl 4-chloro-3-oxobutanoate)制备乙烷-(R)-4-氯-3-羟基丁酸[ethyl-(R)-4-chloro-3-hydroxy butanoate],底物采取批式流加方式,最终底物量达到 215g/L(1.3mol/L),转化一批次后产物的产率达91.7%,ee值达99.6%。
2 双细胞耦合的辅酶再生系统
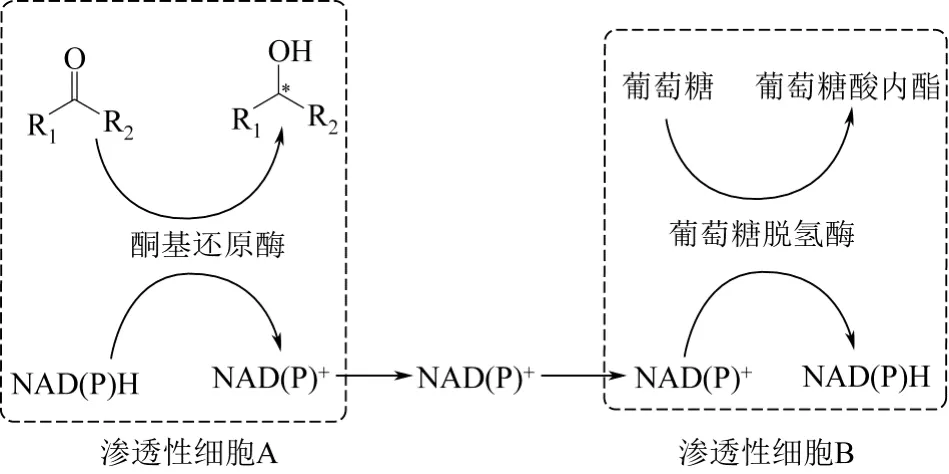
图4 渗透性细胞耦合构建辅酶再生体系
上述的单细胞转化制备手性化合物技术特点是利用细胞固有或人为设计的方式将酶催化转化与辅酶再生酶的催化反应集成在同一细胞内进行,也有学者通过对含有相关耦合酶的细胞进行处理,在不将酶提取出胞外的情况下,直接耦合这两种细胞进行催化转化与辅酶再生反应,即通过再生辅酶在双细胞间来回渗入进出来构建双细胞胞内酶耦合催化体系[28-29]。如Zhang等[30]将两种渗透性细胞A和细胞B耦合,实现NAD(P)H的再生与持续利用(图4)。其基本过程是将表达NADPH依赖型的酮基还原酶(keto reductase,KR)的短小芽胞杆菌(Bacillus pumilusPhe-C3)与表达葡萄糖脱氢酶(GDH)的枯草芽胞杆菌(Bacillus subtilisBGSC 1A1)进行透性化处理后将两细胞耦合培养同时胞外添加一定量的辅酶转化乙烷-3-酮基-4,4,4-三氟醚丁酸(ethyl 3-keto-4,4,4-trifluorobutyrate)生成相应的R型醇盐。在几批次的转化中均表现出了较好的转化稳定性,最终辅酶总转化数(total turn number,TTN)达1620,产物收率与ee值均要优于相应的全细胞催化结果。这种细胞共培养进行转化的方法较简单,耦合条件优化好后也有利于工业化的应用。
Šalić 等[31]在微反应器中固定化渗透化酵母细胞,利用酵母细胞中的 NADH依赖型醇脱氢酶(ADH)还原醛反应实现外源添加的辅酶NAD+的再生与持续供给。这是利用渗透化细胞构建普适性的辅酶再生系统,可以与相当多的辅酶依赖型催化酶的催化反应相耦合,应用于诸如手性化合物的酶催化制备中。在随后的6天测试中,渗透性细胞中的辅酶再生体系表现出了较好的催化稳定性。实验结果证明渗透性细胞在给酶催化提供稳定与可反复多批次利用的氧化还原力方面具有良好的应用可行性。
3 胞外酶耦合辅酶再生系统
胞外酶经提取从细胞内分离出来后再进行催化转化制备手性化合物,同时从另一培养的细胞中提取分离专一性较强的辅酶再生酶与之相耦合在胞外实现催化反应的连续进行。这种胞外酶直接耦合方法具有专一、无副产物、合成效率高、产物纯度高等特点。
3.1 胞外纯酶耦合
大部分的胞外酶一般需经纯化后再进行耦合催化生产,这种体系的优势是反应专一性强、反应机理清楚。根据辅酶再生酶的催化情况分为底物耦合法(一种酶催化两种底物)和双酶耦合法(两种酶分别催化两种底物)[12]。辅酶胞外酶法再生己有许多实验室规模应用的实际例子[32-35]。在工业应用中的一个瓶颈是由于辅酶的小分子特性,辅酶极易从酶耦合体系中泄露出去,因此胞外酶耦合再生方法需解决再生辅酶的留存与反复多批次利用问题。
Rocha-Martín等[36]应用共价固定法将源于极端嗜热菌(Thermus thermophilus)的NAD+专一性再生酶NADH氧化酶(NADH oxidase,NOX)通过其N末端共价固定于带有CNBr的琼脂糖上与同样源于Thermus thermophilus的次级醇脱氢酶(TtADH)相耦合,在固相介质上进行动力学拆分rac-1-苯基乙醇(rac-1-phenylethanol)的反应,共价固定化的NOX持续氧化拆分反应生成的NADH使之不断再生成氧化型辅酶 NAD+,持续为拆分反应提供所需的还原力。这个过程与 NOX的非共价结合相比较能够实现反应的多批次进行。
除了将液相中的催化改变为固相催化来解决再生辅酶反复多批次利用外,Li 等[37]通过将小分子辅酶与纳米粒子相连接的方法增大辅酶的质量与体积,也可实现双酶耦合催化中再生辅酶反复多批次利用。通过对硅纳米颗粒进行活化带上活性羰基基团制备了纳米颗粒固定化的辅酶 NAD+并将其应用于酮基还原酶/甲酸脱氢酶耦合反应中转化丙酮酸生成相应的L型乳酸(图5)。由于辅酶与纳米颗粒固定化相连,除了辅酶具备了纳米颗粒运动特性可加速反应进行外,再生辅酶也可很容易通过离心或过滤等方法回收进行下一批次的利用。实验结果测定纳米颗粒辅酶载量是73mg/g,耦合体系反应6批次后催化活性还能保留60%。
3.2 无细胞抽提液中内源性辅酶再生系统的利用

图5 纳米颗粒固定化辅酶耦合催化体系
上述胞外酶催化辅酶再生法的设计具有很强的针对性,可以利用专一性的辅酶再生酶进行辅酶的再生反应,如氧化型辅酶 NAD(P)+专一性再生酶NAD(P)H氧化酶[38-40],还原型辅酶NAD(P)H专一性再生酶甲酸脱氢酶等[41-43]。但这种酶法再生辅酶在耦合反应过程及工业化应用过程中却可能存在着辅酶再生酶与主反应催化酶酶学反应特性不相适应等问题,大大降低了耦合反应效率。另外,上述胞外酶耦合催化反应一般是用电泳级纯化酶进行构建,在工业应用过程中无疑增加了成本,而且据有些学者研究,纯化后的酶耦合催化效率反而会出现降低的现象[44]。
Yan等[45]利用表达羰基还原酶的重组E.coli的无细胞抽提液转化多种酮基化合物,在此无细胞抽提液加入葡萄糖后发现此体系出现了辅酶 NADPH再生的现象,经分析是无细胞抽提液中含有E.coli细胞内源性表达的NADPH再生系统。这个结果揭示了容易被大多数学者所忽略的无细胞抽提液中本身存在的细胞内源性辅酶再生酶的应用价值。进一步研究发现此无细胞抽提液中还含有不止一种辅酶再生系统,加入海藻糖、果糖、乳糖和芽糖等均有辅酶再生现象,其中加入葡萄糖和海藻糖的耦合催化效率最高。这种利用细胞内源性辅酶再生系统进行辅酶再生实现手性化合物连续转化的方法有时可获得比全细胞催化更高效的反应催化效率,而且与游离纯化酶耦合体系相比较也具有方法简便、不需外加辅酶再生酶等优势。Yang等[46]在利用蔷薇色酵母(Rhodotorulasp.AS2.2241)转化乙酰吡啶和芳香族酮化合物合成相应手性醇化合物过程中也是利用了细胞内源性耦合酶催化系统在胞外实现了手性化合物的生成与辅酶 NADPH的再生,TTN值达4220,手性醇化合物纯度达96%~99%。
4 总结与展望
生物酶催化法制备手性化合物(手性醇或手性胺等)较化学法等具有专一性强,反应效率高,过程简单与环保等突出特点,但大多数需要昂贵的、稳定性差的烟酰胺类小分子辅酶起到传递电子和质子等作用,因此小分子辅酶在酶催化反应中的稳定持续供给反应所需的氧化还原力是手性化合物工业化生产需要重点解决的问题。
上述的辅酶再生工程中采取了代谢调控、基因工程设计或纯酶、无细胞抽提液耦合等方法,以不同的形式解决辅酶的供应问题。这类方法的核心是辅酶本身的存在与再生。若能采取更直接的方式,如筛选或设计能直接利用水分子或氢气做为电子供体的氧化还原酶,将有可能根本上解决辅酶问题[13]。在目前的条件下,则需更加注重辅酶供给体系的构建,其中关键点是再生辅酶在反应体系中的稳定留存问题,尤其是胞外酶耦合催化方法,应采取合适的工艺过程提高再生辅酶的反复与多批次利用的能力,这对辅酶依赖型酶催化的最终反应效率具有重要影响,由此才可真正实现包括手性化合物在内的大批重要化学品的绿色工业化生产过程。
[1]Liu J Y,Lin Y P,Qiu H,et al.Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models[J].European Journal of Pharmaceutical Sciences, 2013,48(4-5):619-627.
[2]Cauda V,Schlossbauer A,Bein T.Bio-degradation study of colloidal mesoporous silica nanoparticles:Effect of surface functionalization with organo-silanes and poly(ethylene glycol)[J].Microporous and Mesoporous Materials,2010,132(9):60-71.
[3]聂尧,徐岩,王海燕,等.重组大肠杆菌不对称还原 2-羟基苯乙酮合成(R)-苯基乙二醇[J].化工进展,2006,25(10):1231-1236.
[4]Patel R N.Microbial-enzymatic synthesis of chiral intermediates for pharmaceuticals[J].Enzyme and Microbial Technology,2002,31:804-826.
[5]Chena B,Dingerdissenb U,Krauterc J G E,et al.New developments in hydrogenation catalysis particularly in synthesis of fine and intermediate chemicals[J].Applied Catalysis A:General,2005,280(1):17-46.
[6]Liu X,Wang Y,Gao H Y,et al.Asymmetric reduction ofα-hydroxy aromatic ketones to chiral aryl vicinal diols using carrot enzymes system[J].Chinese Chemical Letters,2012,23:635-638.
[7]Wang L J,Li C X,Ni Y,et al.Highly efficient synthesis of chiral alcohols with a novel NADH-dependent reductase fromStreptomyces coelicolor[J].Bioresource Technology,2011,102(4):7023-7028.
[8]Alanvert E,Doherty C,Moody T S,et al.Highly stereoselective biocatalytic reduction of alpha-halo ketones[J].Tetrahedron:Asymmetry,2009,20(9):2462-2466.
[9]Ye Q,Yan M,Xu L,et al.A novel carbonyl reductase fromPichia stipitisfor the production of ethyl(S)-4-chloro- 3-hydroxybutanoate[J].Biotechnol.Lett.,2009,31(1):537-542.
[10]羊明,徐岩,穆晓清,等.一种新的高立体选择性羰基还原酶的性质及分离[J].化工进展,2006,25(9):1082-1088.
[11]Zhao H M,van der Donk W A.Regeneration of cofactors for use in biocatalysis[J].Current Opinion in Biotechnology,2003,14:583-589
[12]张翀,邢新会.辅酶再生体系的研究进展[J].生物工程学报,2004,20(6):811-816.
[13]江金鹏,吴旭日,陈依军.解决氧化还原酶反应体系中辅酶问题的策略及其应用[J].生物工程学报,2012,28(4):410-419.
[14]Liu W F,Wang P.Cofactor regeneration for sustainable enzymatic biosynthesis[J].Biotechnology Advances,2007,25:369-384.
[15]van der Donk W A,Zhao H M.Recent developments in pyridine nucleotide regeneration[J].Current Opinion in Biotechnology,2003,14:421-426.
[16]Dirk Weuster-Botz H J.Cofactor regeneration in phototrophic cyanobacteria applied for asymmetric reduction of ketones[J].Appl.Microbiol.Biotechnol.,2007,75(5):1031-1037.
[17]Gao K,Song Q X,Wei D Z.Coupling of enantioselective biooxidation of dl-1,2-propanediol and bioreduction of pinacoloneviaregeneration cycle of coenzyme[J].Appl.Microbiol.Biotechnol.,2006,71:819-823.
[18]Liang Liya,Liu Rongming,Chen Xu,et al.Effects of over expression of NAPRTase,NAMNAT,and NAD synthetase in the NAD(H)biosynthetic pathways on the NAD(H)pool,NADH/NAD+ratio and succinic acid production with different carbon sources by metabolically engineeredEscherichiacoli[J].Biochemical Engineering Journal,2013,81:90-96.
[19]Liang Liya,Liu Rongming,Wang Guangming,et al.Regulation of NAD(H)pool and NADH/NAD+ratio by over expression of nicotinic acid phosphoribosyltransferase for succinic acid production inEscherichia coliNZN111[J].Enzyme and Microbial Technology,2012,51:286- 293.
[20]Schroer K,Luef K P,Hartner F S,et al.Engineering thePichia pastorismethanol oxidation pathway for improved NADH regeneration during whole-cell biotransformation[J].Metabolic Engineering,2010,12:8-17.
[21]Amidjojo M,Weuster-Botz D.Asymmetric synthesis of the chiral synthon ethyl(S)-4-chloro-3-hydroxybutanoate usingLactobacillus kefir[J].Tetrahedron:Asymmetry,2005,16:899-901.
[22]Ye Q,Cao H,Yan M,et al.Construction and co-expression of a polycistronic plasmid encoding carbonyl reductase and glucose dehydrogenase for production of ethyl(S)-4-chloro-3-hydroxybutanoate[J].Bioresource Technology,2010,101:6761-6767.
[23]Jung J,Park S,Kim H K.Synthesis of a chiral alcohol using a rationally designedSaccharomyces cerevisiaereductase and a NADH cofactor regeneration system[J].Journal of Molecular CatalysisB:Enzymatic,2012,84:15-21.
[24]Sun J A,Zhang L Y,Rao B,et al.Enhanced acetoin production bySerratia marcescensH32 with expression of a water-forming NADH oxidase[J].Bioresource Technology,2012,119:94-98.
[25]王普,祖蕾,何军邀,等.基因工程菌在不对称还原制备手性醇中的应用进展[J].化工进展,2008,27(7):977-982.
[26]Bratigama S,Dennewalda D,Schürmann M,et al.Whole-cell biocatalysis:Evaluation of new hydrophobic ionic liquids for efficient asymmetric reduction of prochiral ketones[J].Enzyme and Microbial Technology,2009,45:310-316.
[27]Ni Y,Li C X,Wang L J.Highly stereoselective reduction of prochiral ketones by a bacterial reductase coupled with cofactor regeneration[J].Org.Biomol.Chem.,2011,9:5463-5468.
[28]Jin J Z,Li H,Zhang J.Improved synthesis of (S)-1-phenyl-2-propanol in high concentration with coupled whole cells ofRhodococcus erythropolisandBacillus subtilison preparative scale[J].Appl.Biochem.Biotechnol.,2010,162:2075-2086.
[29]Rundbäck F,Fidanoska M,Adlercreutz P.Coupling of permeabilized cells ofGluconobacter oxydansandRalstonia eutrophafor asymmetric ketone reduction using H2as reductant[J].Journal of Biotechnology,2012,157:154-158.
[30]Zhang J,Witholt B,Li Z.Coupling of permeabilized microorganisms for efficient enantioselective reduction of ketone with cofactor recycling[J].Chem.Commun.,2006(4):398-400.
[31]Šalić A,Faletar P,Zelić B.NAD+regeneration in a microreactor using permeabilized baker’s yeast cells[J].Biochemical Engineering Journal,2013,77:88- 96.
[32]曹政,王亚军,肖黎,等.羰基还原酶不对称还原(R)-6-氰基-5-羟基-3-羰基己酸叔丁酯[J].生物加工过程,2013,11(1):17-22.
[33]Iqbal N,Rudroff F,Brigĕ A.Asymmetric bioreduction of activated carbon-carbon double bonds using Shewanella yellow enzyme(SYE-4)as novel enoate reductase[J].Tetrahedron,2013,68:7619-7623.
[34]Ni Y,Xu J H.Biocatalytic ketone reduction:A green and efficient access to enantiopure alcohols[J].Biotechnology Advances,2012,30:1279-1288.
[35]Stuermer R,Hauer B,Hall M,et al.Asymmetric bioreduction of activated C=C bonds using enoate reductases from the old yellow enzyme family[J].Current Opinion in Chemical Biology,2007,11:203-213
[36]Rocha-Martín J,Vega D,Bolivar J M,et al.New biotechnological perspectives of a NADH oxidase variant fromThermus thermophilusHB27 as NAD+recycling enzyme[J].BMC Biotechnology,2011,11:101-121.
[37]Li Y H,Liang H,Sun L W.et al.Nanoparticle-tethered NAD+with in situ cofactor regeneration[J].Biotechnol.Lett.,2013,35:915-919.
[38]Odman P,Wellbornb W B,Bommarius A S.An enzymatic process to α-ketoglutarate from L-glutamate-the coupled system L-glutamate dehydrogenase-NADH oxidase[J].Tetrahedron:Asymmetry,2004,15:2933-2937.
[39]Talwalkar A,Kailasapathy K,Hourigan J,et al.An improved method for the determination of NADH oxidase in the presence of NADH peroxidase in lactic acid bacteria[J].Journal of Microbiological Methods,2003,52:333- 339.
[40]Findrik Z,Simunovic I,Vasić-Rački Ð.Coenzyme regeneration catalyzed by NADH oxidase fromLactobacillus brevisin the reaction of L-amino acid oxidation[J].Biochemical Engineering Journal,2008,39:319-327.
[41]Labrou N E.Improved purification ofCandida boidiniiformate dehydrogenase[J].Bioseparation,2000,9:99-104.
[42]Bai Y L,Yang S T.Production and separation of formate dehydrogenase fromCandida boidinii[J].Enzyme and Microbial Technology,2007,40:940-946.
[43]Bekhouche M,Doumèche B,Blum L J.Chemical modifications by ionic liquid-inspired cations improve the activity and the stability of formate dehydrogenase in[MMIm][Me2PO4][J].Journal of Molecular Catalysis B:Enzymatic,2010,65:73-78.
[44]Geueke B,Riebel B,Hummel W.NADH oxidase fromLactobacillus brevis- a new catalyst for the regeneration of NAD[J].Enzyme and Microbial Technology,2003,32:205-211.
[45]Yan Z,Nie Y,Xu,et al.Biocatalytic reduction of prochiral aromatic ketones to optically pure alcohols by a coupled enzyme system for cofactor regeneration[J].Tetrahedron Letters,2011,52:999-1002.
[46]Yang W,Xua J H,Pan J,et al.Efficient reduction of aromatic ketones with NADPH regeneration by using crude enzyme from Rhodotorula cells and mannitol as cosubstrate[J].Biochemical Engineering Journal,2008,42(1):1-5.
