差示扫描量热法测定草酸艾司西酞普兰纯度
2014-07-18张颖卓崔秀兰杨庆云吴松
张颖卓,崔秀兰 杨庆云,吴松
差示扫描量热法测定草酸艾司西酞普兰纯度
张颖卓,崔秀兰 杨庆云,吴松
(内蒙古工业大学,呼和浩特 010000) (中国医学科学院药物研究所,北京 100050 )
采用差示扫描量热法(DSC),根据DSC曲线利用纯度分析软件测定草酸艾司西酞普兰的纯度。对实验条件进行了优化,升温速率为4.0 K/min,称样量为2~3.2 mg。测得草酸艾司西酞普兰的纯度为99.17%,相对标准偏差为0.05%(n=6)。该法测定结果与非水滴定法测定结果(99.24%)基本一致。差示扫描量热法可用于测定草酸艾司西酞普兰纯度,方法操作简便、结果准确。
草酸艾司西酞普兰;纯度;差示扫描量热法;非水滴定法
西酞普兰为选择性5-羟色胺再摄取抑制剂,临床用于抑郁症的治疗[1]。其分子式中存在一个手性中心,经药理学研究表明,S-构型体的抗抑郁作用比R-构型体的至少强100倍[2]。2006年草酸艾司西酞普兰(分子式为C20H21FN2O·C2H2O4)在国内上市[3]。目前国内一直没有公认的草酸艾司西酞普兰对照品供应,在进行含量研究时需从国外购买法定对照品,价格较高。
自20世纪60年代起,差示扫描量热法(DSC)已被用于药品的定性和纯度的测定,20世纪80年代逐渐发展成熟,并在药品检测中得到广泛应用[4]。目前美国药典已经把DSC法作为测定药物纯度的方法之一。笔者采用DSC法对草酸艾司西酞普兰的纯度进行了测定,同时用非水滴定法对测定结果的准确性进行验证。采用DSC法测定草酸艾司西酞普兰纯度无需对照品,操作简便,结果准确。
1 实验部分
1.1 主要仪器与试剂
差示扫描量热仪:DSC-1型,瑞士Mettler Toledo公司,;
热重分析仪:TGA/DSC-1型,瑞士Mettler Toledo公司;
电位滴定仪:Titrino Plus 848型,瑞士Metrohm公司;
冰乙酸:分析纯,北京化工厂;高氯酸:分析纯,国药集团化学试剂有限公司;草酸艾司西酞普兰:中国医学科学院药物研究所。
1.2 实验方法
1.2.1 差示扫描量热法
精密称取草酸艾司西酞普兰样品2.0~3.2 g,置于40 μL标准Al2O3坩埚中,压盖,放入差示扫描量热仪中,以40 μL标准Al2O3空坩埚为参比,进行分析,记录DSC曲线。采用Mettler Toledo公司的分析软件计算草酸艾司西酞普兰样品纯度。
1.2.2 非水滴定法
称取草酸艾司西酞普兰样品约0.1 g,精密称定,置于滴定池中,用冰乙酸20 mL溶解。按照电位滴定法,用HClO4滴定液(0.1 mol/L)滴定,并将滴定结果用空白试验校正。以终点时滴定液体积计算含量,每1 mL HClO4滴定液(0.1 mol/L)相当于41.443 mg 草酸艾司西酞普兰。
2 纯度计算公式
实验使用的分析软件(Stare software)来源于Mettler Toledo公司,该软件以Van’t Hoff熔点下降理论[5]为依据,见式(1)。
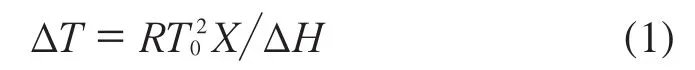
式中:ΔT——熔点降低值,K;
R——气体常数,8.314 J/(K·mol);
T0——纯物质的溶点(凝固点),K;
X——杂质含量(摩尔分数);
ΔH——物质的摩尔熔融焓,J/mol。
由于草酸艾司酞普兰样品中可能含有微量杂质,其主成分的熔点会随杂质含量的提高逐渐降低,可通过修正后的Van’t Hoff方程表示,见式(2)

式中:TS——样品熔化平衡温度,K;
F——已熔化固体的质量分数。
方程(2)中已熔化固体的质量分数F按下式计算:
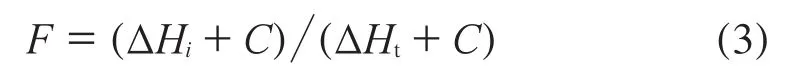
式中:ΔHi——对应于温度Ti时的热流量,J/mol;
Ht——熔化过程中总热流量,J/mol;
C——线性修正参数,C=RT0X/ΔH。
由式(2)可知,TS与1/F呈线性关系,其函数关系图称为1/F图(或Van’t Hoff图),纵坐标截距即为T0,根据式(2)可计算得杂质含量X,从而得到草酸艾司酞普兰纯度结果。
3 结果与讨论
3.1 实验条件优化
分别对溶剂残留、升温速率与称样量3个影响因素进行研究,为草酸艾司西酞普兰纯度测定选择最佳检测条件。
3.1.1 溶剂残留检测
若草酸艾司西酞普兰样品中含有残留溶剂,对其进行差示扫描量热检测时,残留溶剂会在升温过程中吸热,产生吸热峰[6-7],严重影响草酸艾司西酞普兰纯度测定结果的准确性。选择升温范围为40~180℃,草酸艾司西酞普兰供试样品DSC检测曲线见图1。由图1可知,除在151℃有明显的草酸艾司西酞普兰热吸收峰外无其它吸热峰出现。
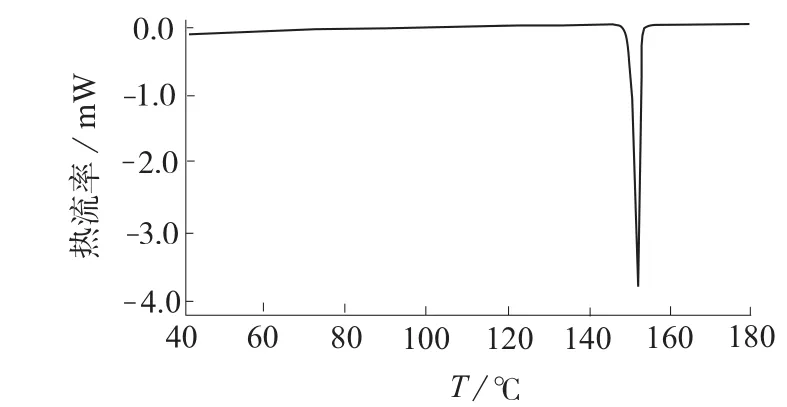
图1 草酸艾司西肽普兰样品DSC曲线
热重分析法(TG)可准确测量样品在温度变化过程中的质量变化及变化速率。以70 μL标准空坩埚为参比,取样品适量,放入热重分析仪中,记录热失重曲线,见图2。从图2中可以看出,样品从40~151℃的升温过程中无质量变化,基线平直。表明草酸艾司西酞普兰样品中无其它残留溶剂存在。
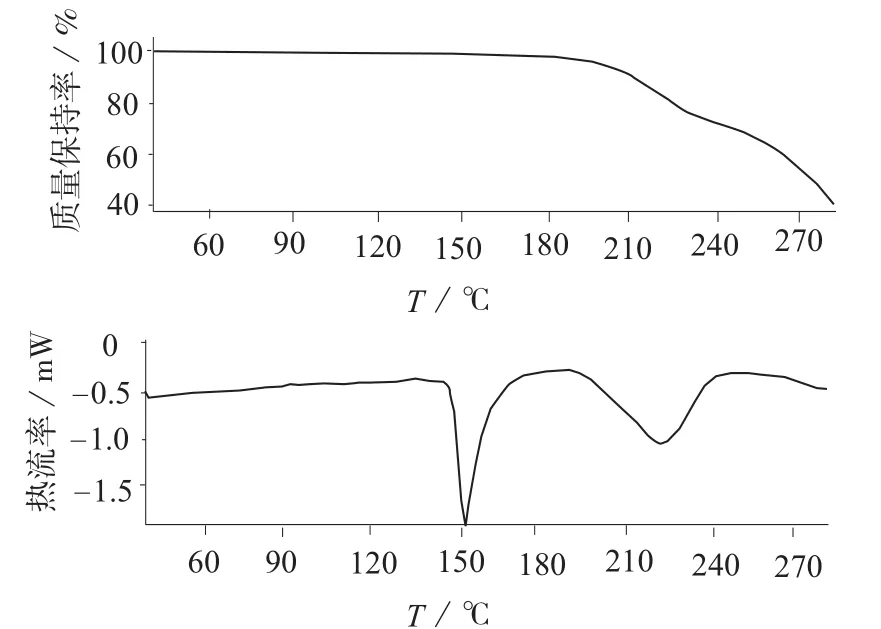
图2 草酸艾司西酞普兰热重分析曲线
3.1.2 升温速率
程序升温速率对DSC曲线的峰温和峰形均产生显著影响,当升温速率增加时,峰温随之升高,峰形变大且更加尖锐。升温速率高会导致样品内部温度分布不均匀,温度梯度增大,吸收峰变宽,使测定结果准确度下降;升温速率低,炉体和样品接近热平衡状态,有利于提高测定结果的准确性,但过低的升温速率会增加实验耗时、仪器负担[8]、产生不光滑DSC检测曲线。
3.1.3 称样量
样品称样量对结果的准确性和灵敏度有一定影响。称样量较少时,可以提高检测灵敏度,消除因升温产生的温度梯度,但称样误差较大[8];称样量较大时,样品内部传热慢、温度梯度大,导致峰形扩大,分辨力下降。
3.1.4 条件优化试验
进行不同水平条件组合下的双因素无重复试验,对结果进行方差分析,选择最佳检测条件。试验设计内容:10个水平条件的升温速率(1,2,3,4,5,6,7,8,9,10 K/min),3个水平条件的样品质量(2.0~2.2,2.5~2.7,3.0~3.2 mg)。试验结果见表1,方差分析结果见表2、表3。
实验结果表明,称样量对草酸艾司西酞普兰纯度测定结果无显著影响(P-value=0.70>0.05),而升温速率对其纯度测定有显著影响(P-value=0.047<0.05),见表2。调整升温速率范围为2~5 K/min,在此范围内升温速率对测定结果无显著影响(P-value=0.18>0.05),见表3。综合分析后最终确定升温速率为4 K/min、称样量为2.0~3.2 mg。
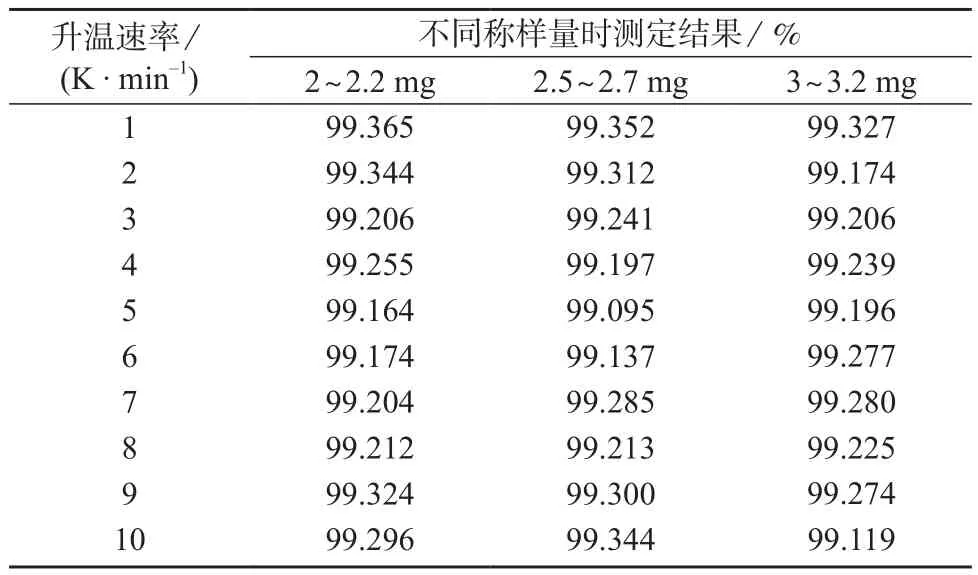
表1 升温速率与称样量双因素无重复试验结果

表2 1~10 K/min升温速率与2~3.2 mg称样量方差分析结果
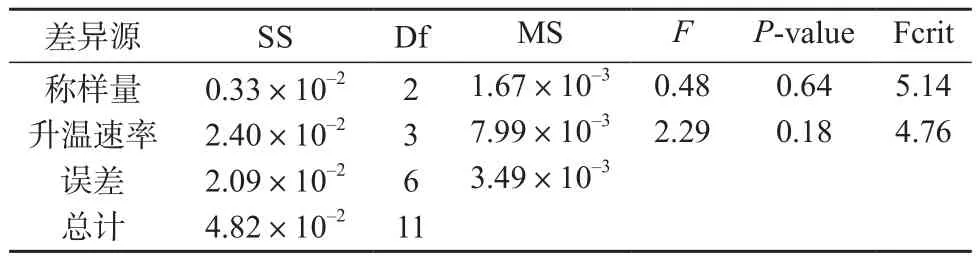
表3 2~5 K/min升温速率与2~3.2 mg称样量方差分析结果
3.2 炽灼残渣的影响
若草酸艾司西酞普兰样品中含有非挥发性无机杂质,则影响测定结果准确性。按照炽灼残渣法(中国药典2010版附录VIIIN),测得残渣量为0.05%。
3.3 方法学线性和精密度考察
分别精密称取6份不同质量(2.0~3.2 mg)的草酸艾司西酞普兰样品,利用差示扫描量热仪对其纯度进行测定,升温速率为4 K/min,记录DSC曲线,见图3。草酸艾司西酞普兰纯度测定结果的平均值为99.23%,相对标准偏差为0.05%,实验结果如见表4。因样品检查炽灼残渣结果为0.05%,所以DSC测定结果应校正为99.17%。以草酸艾司西酞普兰的称样量为横坐标(x),测得的吸热峰面积为纵坐标(y),绘制标准曲线,得曲线方程为y=84.613x+26.289,相关系数r=0.999 9,见图4。综合上述分析可知,测定草酸艾司西酞普兰纯度的DSC法具有良好的线性和精密度。

图3 差示扫描量热法(DSC)检测草酸艾司西酞普兰样品分析图

表4 DSC法测定草酸西肽普兰纯度实验结果

图4 草酸艾司西酞普兰标准曲线
3.4 非水滴定法测定结果
非水滴定法是在水以外的溶剂中进行滴定的方法。非水溶剂可增大样品的溶解度,增强其酸碱度,扩大酸碱滴定范围,得到明显的终点突跃。此法具有灵敏准确、选择性高等优点,可准确测定草酸艾司西酞普兰纯度,因此用该法验证差示扫描量热法测定结果的准确性。
采用非水滴定法分别对6份草酸艾司西酞普兰样品进行纯度检测,检测结果分别为99.15%,99.18%,99.22%,99.27%,99.29%,99.31%,平均值为99.24%,相对标准偏差为0.06%。由此可见,差示扫描量热法与非水滴定法对草酸艾司西酞普兰纯度测定结果基本一致。
4 结论
(1)差示扫描量热法可在程序升温的条件下测量样品与参比物之间的热量差随温度的变化情况。在进行物质纯度检测的过程中,DSC可对供试样品产生的热效应给予及时补偿,使供试样品与参比物之间无温差、无热交换,供试样品的升温速率始终跟随炉温进行线性升温,使检测方法有较高的灵敏度和精确度。
(2)采用差示扫描量热法测定草酸艾司西酞普兰纯度具有试样用量少,操作简单,无污染,灵敏度、精密度高等优点。该方法可准确检测纯度在98%以上的样品,在检测过程中无需使用对照品[9]。经非水滴定法验证,差示扫描量热法测定草酸艾司西酞普兰纯度是可行性的,为草酸艾司西酞普兰纯度测定提供了一种新的检测方法。
[1] 杜瑜,李焕德.抗抑郁新药西酞普兰的药代动力学[J].中国临床药理学杂志,2005,21(4): 307-310.
[2] Sorbera L A,Revel L,Martin L,el al. Escitalopram oxalate[J]. Drugs Future,2001,26(2): 115-120.
[3] 乌兰辉,吴松,卢建勋,等.反相高效液相色谱法拆分西酞普兰对映异构体[J].药物分析杂志,2012,32(1): 71-73.
[4] 纪雷,王岩.差热分析法测定化工品、药品纯度[J].化学分析计量,2001,10(6): 23-24.
[5] 蒙根,朱海燕.差示扫描量热法测定马来酸酐纯度[J].化学分析计量,2008,17(5): 41-42.
[6] Giron D,Foldbronn C. Place of DSC purity analysis in pharmaceutical development[J]. Journal of Thermal Analysis,1995,44: 217-251.
[7] Yi Li,Pui Shan Chow. Quantification of polymorphic impurity in an enantiortopic polymorph system using differential scanning calorimetry, X-ray powder diffraction and Raman Spectroscopy[J]. International Journal of Pharmaceutics,2011,415: 110-118.
[8] 孙颖. DSC热分析技术建立结晶有机化合物纯度分析方法的研究[J].广西化工,2002,31(2): 29-31.
[9] 陈青.差示扫描量热法单峰测定物质的纯度[J].分析仪器,2005(3): 42-46.
Determination of Escitaliporam Oxalate Purity by Differential Scanning Calorimetry Method
Zhang Yingzhuo, Cui Xiulan
(Inner Mongolia University of Technology, Hohehot 010000, China)
Yang Qingyun, Wu Song
(Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China)
The differential scanning calorimetry (DSC) technique was used to determine the purity of escitalopram oxalate, according to the DSC curve and by using the purity analysis software. The experimental conditions were optimized, the heating rate was 4.0 K/min and the sample mass was 2-3.2 mg. The purity result determined by DSC method was 99.17%, RSD was 0.05%(n=6). The result was consistent with the result (99.24%) determined by the non-aquseous trirtation method. The DSC method is accurate and simple for the determination of escitalopram oxalate purity.
escitalopram oxalate; purity; differetial scanning calorimetry; non-aqueous trirtation method
O657.99
A
1008-6145(2014)01-0057-04
联系人:张颖卓;Yingzhuo99@aliyun.com
2013-11-05
10.3969/j.issn.1008-6145.2014.01.016
