不同采收季节的淫羊藿中朝藿定C和淫羊藿苷的含量比较
2014-07-16左建平安显辉
左建平,安显辉
(珠海星光制药有限公司,广东 珠海 519180)
淫羊藿为小檗科植物淫羊藿 Epimedium brevicornum Maxim.、箭叶淫羊藿 Epimedium sagittatum(Sieb.et Zucc.)Maxim.、柔毛淫羊藿 Epimedium pubescens Maxim.、巫山淫羊藿 Epimedium wushanense T.S.Ying、或朝鲜淫羊藿 Epimedium koreanum Nakai的干燥叶[1]。另外粗毛淫羊藿 E.acuminutum Franch.、天平山淫羊藿E myrianthum Steam和黔岭淫羊藿 E.leptorrhizum Steam为《贵州省中药材、民族药材质量标准》允许使用品种,淫羊藿属植物在中国约有40余种[2]。淫羊藿 Herba Epimedii有益精气、坚筋骨、补腰膝、强心力之功效。迄今已报道的淫羊藿属植物约55种[3],广泛分布于除新疆、西藏、内蒙古、河北、山东以外的各个省份。贵州是全国淫羊藿药材产量最大的地区,主流品种为粗毛淫羊藿,其次为黔岭淫羊藿、巫山淫羊藿。本研究通过对粗毛、黔岭、巫山淫羊藿药材的不同采收季节、不同产地中淫羊藿苷和朝藿定C的含量进行测定,为贵州产主流淫羊藿药材的质量评价提供科学依据,为含淫羊藿药材的中成药质量控制提供参考。
1 仪器与试药
HP1100型高效液相色谱仪 (在线脱气机、四元泵、自动进样器、柱温箱、VWD检测器,ChemStation色谱工作站);KQ-300DA型超声波清洗器(昆山市超声仪器有限公司)。朝藿定C对照品(自制并鉴定结构,经高效液相色谱法检测纯度大于98%);淫羊藿苷(中国药品生物制品检定所,批号为0737-9910);乙腈为色谱纯,水为乐百氏纯净水,其他试剂均为分析纯;粗毛淫羊藿、巫山淫羊藿(均自采于贵州省贵阳市修文县、贵州省贵阳市药用植物园),黔岭淫羊藿(自采于贵州省安顺市关岭自治县)。
2 方法与结果
2.1 色谱条件
色谱柱:SinoChrom ODS-AP 柱(250 mm × 4.6 mm,5.0 μm,大连依利特分析仪器有限公司 );柱温:25℃;流动相:乙腈(A)-水(B),梯度洗脱(0 ~22 min,27% →29%A;22 ~23 min,29% →100%A;23~34 min,100%A;34~36 min,100% →27%A;36~50 min,27%A);流速:1 mL /min;进样量:10 μL;检测波长:270 nm。
2.2 溶液制备
精密称取朝藿定C和淫羊藿苷对照品,加适量甲醇制成朝藿定 C和淫羊藿苷质量浓度分别为1.322,1.513 g/L的混合溶液,摇匀,即得对照品溶液。分别取不同月份的粗毛淫羊藿、巫山淫羊藿、黔岭淫羊藿粉末(过80目筛)0.2 g(同时另取粗毛淫羊藿、巫山淫羊藿、黔岭淫羊藿药材粉末测定水分),精密称定,置锥形瓶中,加70%乙醇20 mL超声提取1 h,并补足减失的质量,用微孔滤膜(0.45 μm)滤过,取续滤液,即得供试品溶液。
2.3 方法学考察
系统适用性试验:分别精密吸取朝藿定C、淫羊藿苷的对照品溶液和混合对照品溶液及3种供试品溶液各10 μL,注入高效液相色谱仪,记录色图谱。结果发现,朝藿定C对照品在14.28 min时出峰,淫羊藿苷对照品在16.89 min时出峰,供试品图谱在相同时间出现和对照品同样的峰,且基线较平,杂质峰较少,且朝藿定C、淫羊藿苷峰和相近的杂质峰分离完全(分离度大于1.5)。见图1。
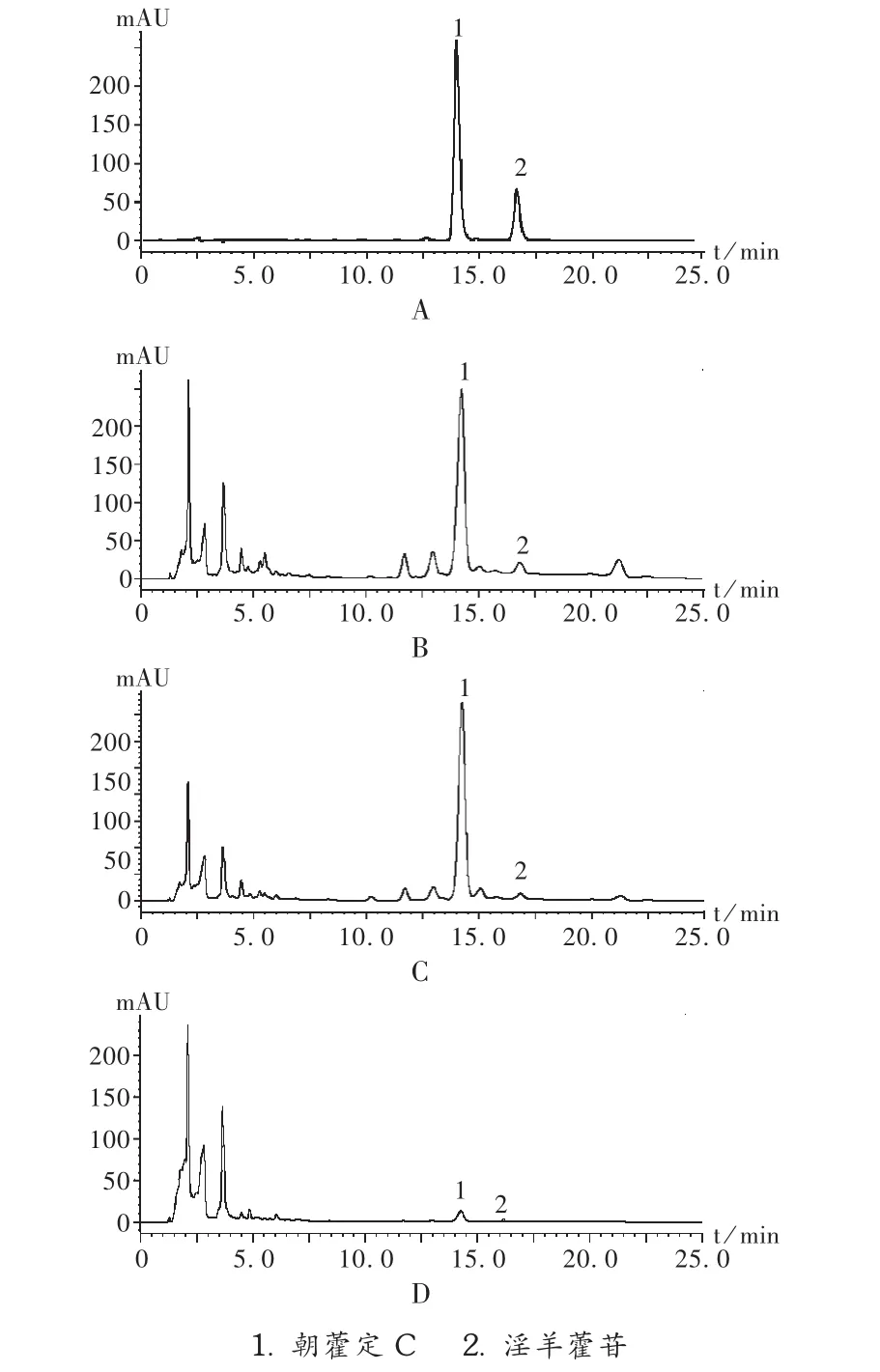
图1 高效液相色谱图
线性关系考察:精密称取干燥的朝藿定C对照品(干燥品按98.0% 计)13.49 mg 和淫羊藿苷对照品 15.13 mg,置 10 mL 容量瓶中,用甲醇溶解并稀释至刻度,摇匀,即得朝藿定C和淫羊藿苷对照品溶液(每1 mL含朝藿定 C 1.322 mg,淫羊藿苷1.513 mg)。精密吸取朝藿定 C对照品溶液1,2,3 mL和淫羊藿苷对照品溶液 0.2,0.4,1 mL,分别置 10 mL 1,2,3 号容量瓶中,再于 1 号瓶中取 1,2,4 mL分别置 4,5,6号 10 mL容量瓶中,并加甲醇定容至刻度,摇匀。分别精密吸取上述6种溶液各10 μL注入高效液相色谱仪,记录色谱,以峰面积(A)为纵坐标、进样量(X)为横坐标进行线性回归,得回归方程,朝藿定 C为 A=18 025.706 4 X+6.255 671 1(r=0.999 9), 淫 羊 藿 苷 为 A=22 664.828 3 X-3.752 616 4(r=0.999 9)。结果表明,朝藿定 C 和淫羊藿苷进样量在 13.22 ~ 396.6 μg 和 3.026 ~151.3 μg 范围内与峰面积呈良好线性关系。
精密度考察:精密量取朝藿定C质量浓度为1.322 g/L的对照品溶液1 mL和淫羊藿苷质量浓度为1.513 g/L的标准溶液0.2 mL,分别置10 mL容量瓶中,加甲醇稀释至刻度,摇匀,即得朝藿定 C质量浓度为 132.2 μg/mL和淫羊藿苷质量浓度为30.26 μg /mL 的溶液,精密吸取 10 μL,注入高效液相色谱仪,重复进样6次,测定峰面积。结果朝藿定C的 RSD为0.43%,淫羊藿苷的 RSD为0.49%,表明仪器精密度良好。
稳定性试验:依法制备供试品溶液,精密吸取10 μL,分别于0,6,8,12,14 h时注入高效液相色谱仪,测定峰面积。结果朝藿定C峰面积的 RSD为0.91%,淫羊藿苷峰面积的 RSD为1.05%,表明供试品溶液在14 h内稳定。
重复性试验:取粗毛淫羊藿药材干燥样品,依法制备5份供试品溶液,精密吸取10 μL,注入高效液相色谱仪,测定峰面积,计算朝藿定C和淫羊藿苷的平均含量。结果朝藿定C和淫羊藿苷的平均含量的 RSD分别为0.84%和1.47%,表明该方法重现性良好。
加样回收试验:精密称取已测定含量的样品 (朝藿定C含量为 0.930 64% ,淫羊藿苷含量为 0.153 18%)0.1 g,置锥形瓶中,分别按80%,100%,120%的比例加入已知质量浓度的对照品混合溶液,每个浓度平行3份,共9份,加70%乙醇20 mL超声提取 1 h,放冷,用 70%乙醇补足减失的质量,用 0.45 μm滤膜过滤,取续滤液,精密吸取10 μL,注入高效液相色谱仪,测定峰面积。结果见表1。
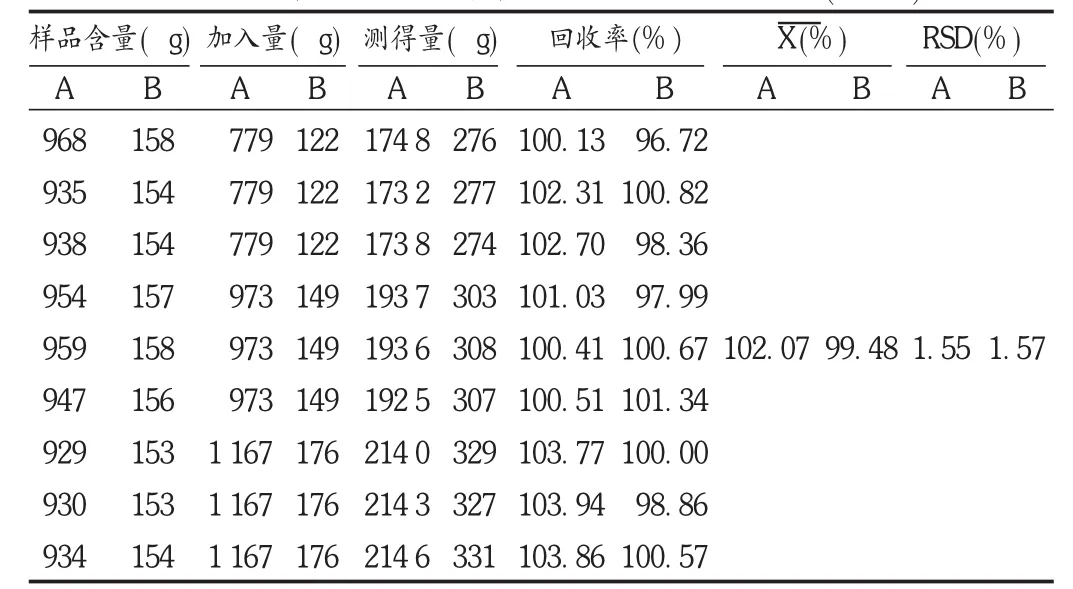
表1 朝藿定C和淫羊藿苷加样回收试验结果(n=9)
2.4 样品含量测定
将于不同月份采集的淫羊藿药材的干燥粉末于80℃干燥3 h,依法制备供试品溶液和对照品溶液,分别进样10 μL,记录色谱图,测定峰面积,按公式计算含量。结果见表2。
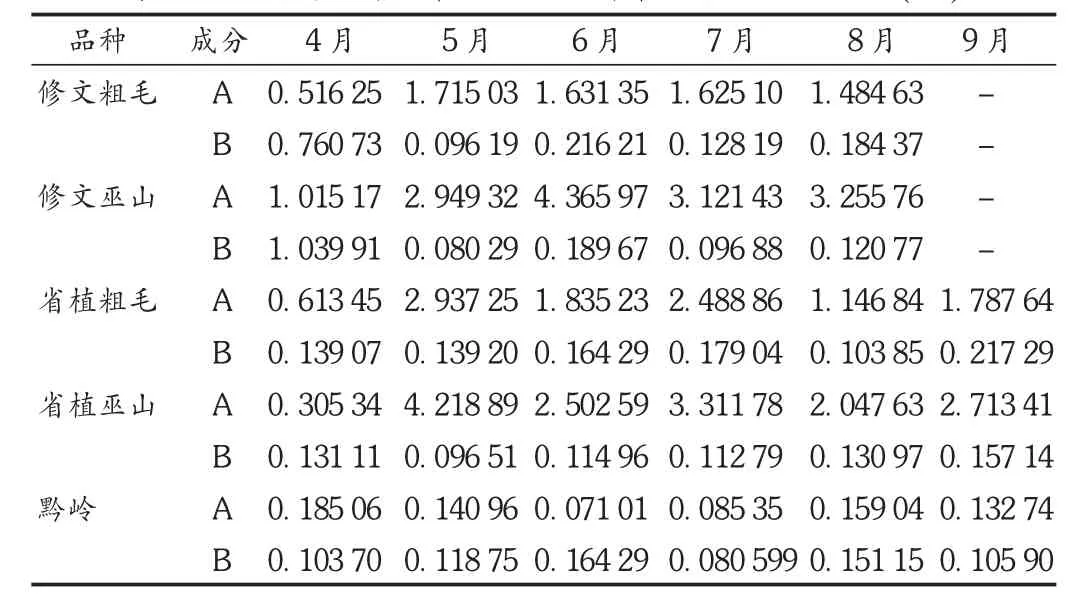
表2 3种样品中朝藿定C和淫羊藿苷含量测定结果(%)
3 讨论
分别考察乙腈-水(28∶72)、乙腈-水(30∶70)及不同乙腈比例在22,24,25 min内从27%到29%的3种梯度洗脱条件,结果以本试验中采用的分离条件较好,朝霍定C和淫羊藿苷能有效分离。淫羊藿药材的主要有效成分为黄酮类化合物,其中朝霍定C、淫羊藿苷在药材中占比较大,而2010年版《中国药典(一部)》仅以淫羊藿苷作为药材的含量测定指标,不能客观反映淫羊藿药材的内在质量,应以朝霍定C和淫羊藿苷作为淫羊藿药材的质控指标。本方法简便、快速、准确、重复性好,能较全面地控制淫羊藿药材中有效成分的含量,应增加淫羊霍苷和朝霍定C作为淫羊藿药材的质控指标。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:306.
[2]王悦云,徐文芬,何顺志.中国淫羊藿小花类群的研究现状[J].贵阳中医学院学报,2005,27(4):6.
[3]郭宝林,肖培根.中药淫羊藿主要种类评述[J].中国中药杂志,2003,28(4):303.
