富镓GanAs团簇稳定性及缺陷特性的密度泛函理论研究
2014-07-13马德明乔红波李恩玲马优恒
马德明,乔红波,李恩玲,施 卫,马优恒
(1.西安理工大学应用物理系,西安710054;2.西安应用光学研究所,西安710065)
1 引 言
Ⅲ-Ⅴ族化合物半导体材料在微电子学和超高速光电子学等领域有着广泛的应用[1],人们用基于密度泛函理论的第一性原理对Ⅲ-Ⅴ族化合物半导体材料在理论计算方面做了大量的研究[2,3],其中对于砷化镓团簇的理论研究也有很多.Balasubramaruan等人较早地研究了Ga2As2团簇的结构[4],Song 等人用分子轨道动力学方法研究了Ga4As4中性团簇的三个具有Td、Ci、D2d对称性的稳定结构[5],Lou 等用基于密度泛函理论的第一性原理研究了Ga5As5团簇结构,认为其基态结构为带四帽的三棱柱结构[6],Vasiliev等在局域密度近似框架下用随时间变化的密度泛函理论研究了Ga4As4和Ga5As5团簇结构的吸收谱[7],赵卫等人用全势能线性糕模轨道分子动力学方法(FD-LMTO-MD)研究了GanAsn(n=5,6,8)表现出半导体性质[8],Gutsev等人用DFT-GGA 方法对GanAsn(n=2~16)系列团簇进行了研究[9],Lu等人利用密度泛函理论研究了GanAsn(3≤n≤14)团簇的环状结构[10],Karamanis等人采用基于密度泛函理论的第一性原理等方法研究了GanAsn(n=2~9)团簇系列的结构和稳定性规律,并对团簇的极化率进行了计算分析[11],杨建宋和李宝兴等采用第一性原理研究了Ga7As7团簇的稳定结构以及相应电荷对其结构的影响[12].这些研究主要集中于砷和镓原子数相等的团簇,对富砷和富镓砷化镓团簇的研究相对较少.砷容易挥发,砷化镓体材料中不可避免的存在着富镓砷化镓团簇缺陷,所以研究富镓砷化镓团簇及其缺陷结构与特性,对砷化镓材料的应用有着重要的意义.人们通过理论计算等手段,对SI-GaAs中的EL 能级可能的微观构型进行了相应分析,常见的是EL2(Ec-0.82 eV)和EL6(Ec-0.38eV)深能级缺陷.到目前为止,已经提出了多种结构模型,认为EL2的可能构型为AsGaVAsVGa,AsGaAsi等,EL6的可能构型为VAsVGa,AsGaVAs等,这些缺陷在材料中以团簇的形态存在[13,14].在前期研究富砷团簇的基础上[15,16],用基于密度泛函理论的第一性原理对富镓中性GanAs(n=1~9)团簇的结构稳定性以及砷化镓材料VAsVGa缺陷的特性进行了研究,以其为砷化镓材料的团簇缺陷研究提供帮助.
2 计算方法
通过基于密度泛函理论(DFT)的第一性原理计 算 软 件VASP(Vienna Ab-initio Simulation Package)完成计算.计算中采用了半局部Perdew-Burke-Ernzerhof(PBE)交换关联泛函和Heyd-Scuseria-Erazerhof(HSE)杂化交换关联泛函,所使用的赝势是投影缀加波赝势Projector Augmented-Wave(PAW),在杂化泛函中有25%的非局部Hartree-Fock交换泛函和75%的半局部交换泛函,筛选参数为0.2 Å-1,平面波截断能为300 eV,总能计算的收敛标准为1×10-6eV,格点分割采用以Γ 点为对称中心的3×3×3 的Monkhorst-Pack方案,并采用Gaussian smearing方法,展宽σ为0.1eV.
3 结果与讨论
3.1 几何结构
在富镓中性GanAs(n=1~9)团簇中所选取的Ga-As键长为2.4Å 至3.7Å 之间,Ga-Ga键长为3.9Å 至5.7 Å 之间,根据前期研究基础,每个团簇设计了各种不同的结构,通过计算分析得到9种不同原子总数的基态结构如图1 所示,其中深色球和浅色球分别代表Ga原子和As原子,相应团簇基态结构的对称性、键长如表1 所示.
3.2 结构稳定性
团簇结合能的二阶差分值定义如下:

式(1)中,DEb(N+1)和DEb(N)为团簇结合能的一阶差分值,D2Eb(N)为团簇结合能的二阶差分值,D2Eb(N)越大,相应团簇越稳定.团簇各基态结构的D2Eb(N)随总原子数的变化关系如图2所示.
从图2中可以得出,随着总原子数的增大,基态团簇的D2Eb(N)呈奇偶交替的变化规律,其总原子数为奇数的团簇比总原子数为偶数的团簇D2Eb(N)大,即总原子数为奇数的团簇比总原子数为偶数的团簇稳定性好.
3.3 化学稳定性
能隙差(Egap)的大小可以反映电子从最高占据轨道向最低空轨道发生跃迁的能力,即团簇参与化学反应的能力,也即团簇的化学稳定性.对GanAs(n=1~9)团簇的各基态结构进行了计算,其Egap随总原子数的变化关系如图3所示.
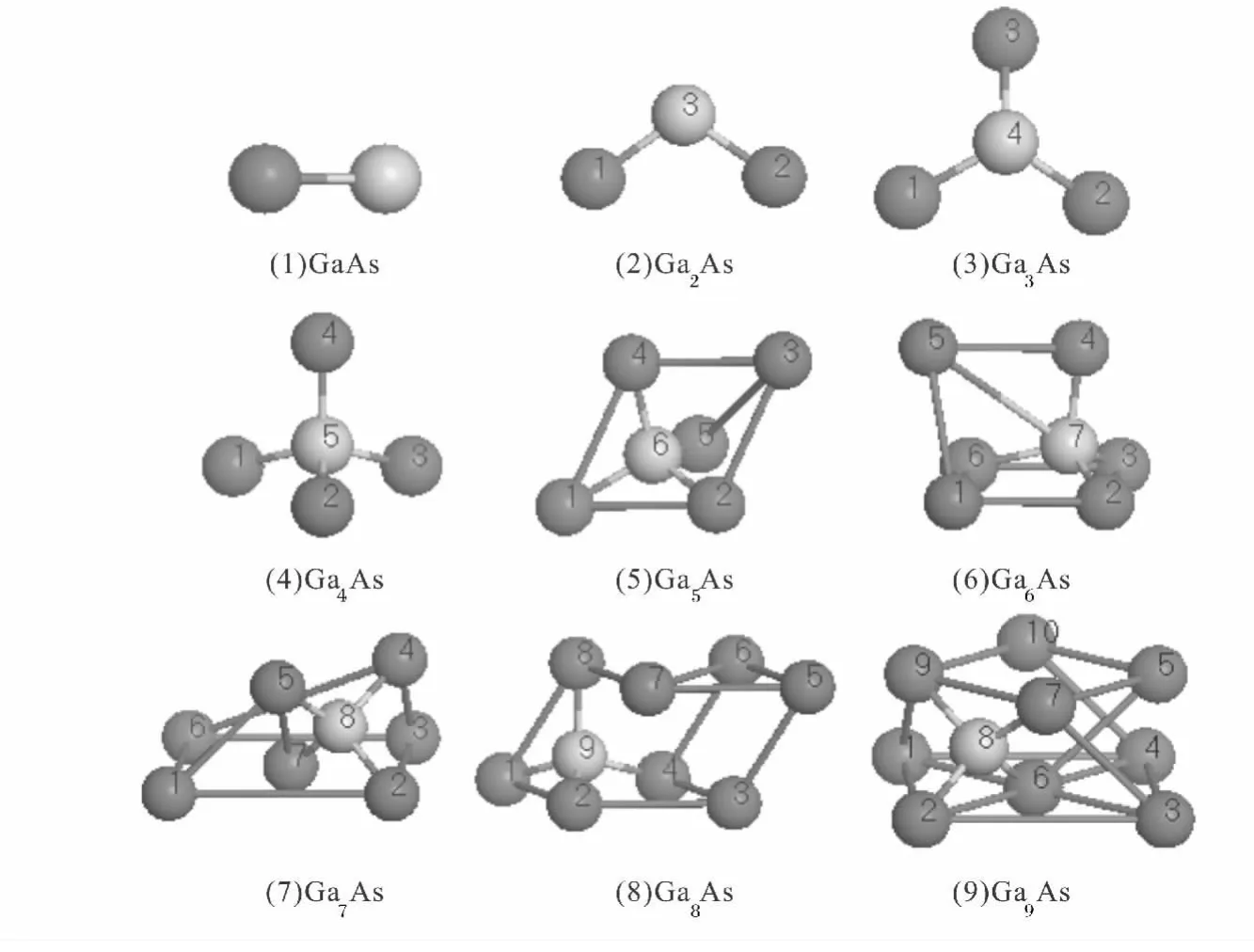
图1 富镓中性GanAs(n=1~9)团簇的基态几何构型Fig.1 Ground-state geometric structures of Ga-rich neutral GanAs(n=1~9)clusters

图2 D2Eb(N)与总原子个数的关系Fig.2 D2Eb(N)as a function of the total atomic numbers
从图3中可以看出,富镓GanAs(n=1~9)团簇的Egap随总原子数的增大呈奇偶交替的变化规律,其中总原子数为奇数的团簇比总原子数为偶数的团簇Egap大,即总原子数为奇数的团簇比总原子数为偶数的团簇稳定;各基态团簇的Egap均在0.06~2.38eV 之间,具有半导体特性;GaAs团簇的Egap为0.891eV,总原子数大于5的富镓砷化镓团簇的Egap也相对较小,小于砷化镓体材料禁带宽度1.43eV,位于禁带较深区域,可以构成砷化镓材料的EL 深能级缺陷,对砷化镓材料的能带结构和光电特性有着重要的影响.

图3 Egap与总原子个数的关系Fig.3 Egapas a function of the total atomic numbers
3.4 振动频率
表2给出了GanAs(n=1~9)团簇基态结构的振动频率.其中最小的振动频率可以反应所得到的结构是否存在虚频,最高振动频率可以反应红外光谱中最强吸收峰的位置,从而判断优化结构的稳定性.
从表2可以看出,所得的振动频率均为正值,表明各结构均为势能面上的极小点,保证了能量的二阶导数矩阵的本征值为正值,相应的结构为平衡结构.这些团簇的振动频率均在THz频段,因此为团簇的THz波检测实验提供了依据,同时也为砷化镓材料的THz波辐射研究提供了一定的帮助.

表1 富镓中性GanAs(n=1~9)团簇的基态几何参数Table 1 Optimized geometric parameters for ground-state structures of Ga-rich neutral GanAs(n=1~9)clusters

表2 富镓中性GanAs(n=1~9)团簇的基态结构的振动频率Table 2 Vibrational frequencies of the ground states structures of Ga-rich neutral GanAs(n=1~9)clusters
3.5 VAsVGa团簇缺陷特性
从以上团簇结构计算可知,GaAs团簇的Egap为0.891eV,可以构成砷化镓材料的深能级缺陷,其可能的结构形态有VAsVGa、AsGaGaAs等,本文对含有VAsVGa缺陷的砷化镓晶体材料进行理论模拟计算,得到相应的能带结构和电子态密度如下图4所示.

图4 VAsVGa缺陷的能带结构和总电子态密度Fig.4 The band structure and the total DOS of VAsVGadefects
从图4中可以看出,由于双空位VAsVGa缺陷的存在,在价带顶部以上出现了三条受主缺陷能级(黑色虚线所示),在导带底部以下出现了四条施主缺陷能级,与缺陷态密度图中所出现的新缺陷峰相对应(黑色粗实线所示),导致布里渊区中心Γ点处的直接带隙宽度减小,并且由该缺陷所造成的最低施主缺陷能级位于导带底以下0.39 eV,该值接近于EL6缺陷能级的实验值(Ec-0.38 eV),为SI-GaAs材料吸收长波限以外的激光提供了依据.
4 结 语
利用基于DFT 的第一性原理对富镓GanAs(n=1~9)团簇结构稳定性及VAsVGa缺陷特性进行了研究.随着总原子数的增大,团簇基态结构的结合能二阶差分值D2Eb(N)和团簇能隙差Egap呈奇偶交替的变化规律,其总原子数为奇数的团簇D2Eb(N)和Egap相对较大,即总原子数为奇数的团簇比总原子数为偶数的团簇稳定.富镓GanAs(n=1~9)团簇各基态结构的Egap均在0.06~2.38eV 之间,具有半导体的特性,GaAs团簇的Egap为0.891eV,总原子数大于5 的富镓砷化镓团簇的Egap也相对较小,小于砷化镓体材料禁带宽度1.43eV,位于禁带较深区域,可以构成砷化镓材料的EL 深能级缺陷,对砷化镓材料的能带结构和光电特性有着重要的影响.VAsVGa缺陷导致砷化镓材料布里渊区中心Γ 点处的直接带隙宽度减小,VAsVGa缺陷施主能级位于导带底以下0.39eV,该值接近于EL6缺陷能级的实验值(Ec-0.38eV),为SI-GaAs材料吸收长波限以外的激光提供了依据.富镓砷化镓团簇的振动频率均在THz频段,为团簇的THz波检测实验提供了依据,同时也为砷化镓材料的THz波辐射研究提供了一定的帮助.
[1] Jin M,Fang Y Z,Shen H,et al.Mechanical property evaluation of GaAs crystal for solar cell[J].Chin.Phys.Lett.,2011,28(8):086101.
[2] Guo Y K,Li E L,Yang D Q,et al.The geometric structure and stability of (GaP)n,(GaP)n+and(GaP)n-(n=1~6)Clusters [J].J.At.Mol.Phys.(原子与分子物理学报),2007,24(1):91.
[3] Li E L,Chen G C,Wang X W,et al.Ab initio investigation of structures and stability of GanNmclusters[J].J.At.Mol.Phys.(原 子 与 分 子 物 理 学报),2007,24(3):244.
[4] Balasubramanian K.Electronic structure of(GaAs)2[J].Chem.Phys.Lett.,1990,171(1-2):58.
[5] Song K M,Ray A K,Khowash P K.On the electronic structures of GaAs clusters[J].J.Phys.B,1994,27(8):1637.
[6] Lou L,Wang L,Chibante L P F,et al.Electronic structure of small GaAs clusters [J].J.Chem.Phys.,1991,94(12):8015.
[7] Vasiliev L,Ogilt S,Chelikowsky J R.Ab initio absorption spectra of gallium arsenide clusters[J].Phys.Rev.B,1999,60(12):8477.
[8] Zhao W,Cao P L,Li B X,et al.Study of the stable structures of Ga4As4cluster using FP-LMTO MD method[J].Phys.Rev.B,2000,62(24):17138.
[9] Gutsev G L,Saha B C.Optical properties of(GaAs)nclusters(n=2~16)[J].J.Phys.Chem.A,2008,112(43):10728.
[10] Lu Q L,Jiang J C,Wan J G,et al.Density-functional study of ring-like GanAsn(3≤n≤14)clusters[J].J.Mol.Stru.:Theochem.,2008,851(1-3):271.
[11] Karamanis P,Begue D,Pouchan C.Structure and polarizability of small(GaAs)nclusters(n=2~6 and 8)[J].Comput.Lett.,2006,2(4):255.
[12] Yang J S,Li B X.First-principle study of Ga7As7ionic cluster and influence of multi-charge on its structure [J].Chin.Phys.B,2010,19(9):097103.
[13] Zou Y X,Wang G Y.Comment on“Atomic model for the EL2defect in GaAs”[J].Phys.Rev.B,1988,38(15):10953.
[14] Zhao Q F,Schlesinger T E,Milnes A G.Evidence for EL6acting as a dominant recombination center in n-type horizontal bridgman GaAs[J].J.Appl.Phys.,1987,61(11):5047.
[15] Ma D M,Li E L,Shi W,et al.Structures and stability of small GamAsnclusters[J].J.At.Mol.Phys.,2008,25(4):984(in Chinese)[马德明,李恩玲,施卫,等.密度泛函理论对GamAsn团簇的结构及稳定性的研究[J].原子与分子物理学报,2008,25(4):984]
[16] Ma D M,Shi W,Li E L,et al.Study on structure and photoelectron spectroscopy of Ga2Asnion clusters[J].Acta Opt.Sin.,2009,29(4):1032(in Chinese)[马德明,施卫,李恩玲,等.Ga2Asn离子团簇结构及其光电子能谱研究[J].光学学报,2009,29(4):1032]
