1,4-二氯蒽醌水解制备1,4-二羟基蒽醌
2014-07-05王贵城段正康颜志祥
王贵城,段正康,颜志祥
(湘潭大学化工学院,湖南 湘潭 411105)
1,4-二氯蒽醌水解制备1,4-二羟基蒽醌
王贵城,段正康,颜志祥
(湘潭大学化工学院,湖南 湘潭 411105)
以廉价的对二氯苯和邻苯二甲酸酐为原料制得1,4-二氯蒽醌,经硼酸催化水解得到1,4-二羟基蒽醌。系统地考察了反应温度、硫酸浓度、原料摩尔比和反应时间对氯转羟基反应的影响。确定了氯转羟基反应制备1,4-二羟基蒽醌的优化实验条件为:以95%浓硫酸为溶剂,在反应温度为220℃、硫酸∶1,4-二氯蒽醌∶硼酸(摩尔比)为37.5∶1.0∶1.4、反应时间为60min的条件下,以1,4-二氯蒽醌计的产物收率为71.0%。探讨和解释了硼酸催化作用下氯代蒽醌转换成羟基蒽醌可能的机理。
1,4-二氯蒽醌;合成;氯转羟基;机理
1,4-二羟基蒽醌属于蒽醌类衍生物,是一种重要的化学中间体,可作为制造还原染料、分散染料及活性染料的中间体,广泛应用于各种染料的合成中,在染料工业中具有重要的地位[1-4]。也有报道[5]1,4-二羟基蒽醌是一种有效的重组人蛋白激酶CK2的抑制剂,能明显抑制蛋白激酶CK2全酶的活性,可以用作抗肿瘤药应用于临床治疗。但是,天然1,4-二羟基蒽醌在植物中的含量非常低[6],因此目前主要以有机合成的方法获取1,4-二羟基蒽醌。
目前国内主要采用对苯二酚路线和对氯苯酚路线[7-11]合成1,4-二羟基蒽醌。但是对苯二酚原料价格高,且对苯二酚有毒,易氧化,高温受热可分解释放出有毒气体;对氯苯酚路线工业生产环境恶劣,且对氯苯酚有强烈的不愉快刺激气味,熔点低,易挥发,易氧化。其他的合成路线由于操作条件苛刻或收率不高等原因未见有工业化生产报道。如El’tsov等[12]在微波条件下用苯酐和对氯苯酚合成1,4-二羟基蒽醌,仅需10min反应完成,但其成本高,不适合工业化生产。Krapcho等[13]用1-氟4-羟基蒽醌在NaOSi(CH3)3和THF的作用下合成1,4-二羟基蒽醌,收率达80%,但原料不易得,反应时间长(大于18h)。
以价廉易得的对二氯苯和苯酐为原料合成1,4-二氯蒽醌,再经硼酸催化水解得到1,4-二羟基蒽醌的方法简单易操作,其关键在于氯基的羟基化。杨慧慧等[14]采用La2O3催化剂在高温高压条件下水解间二氯苯制备间二苯酚,但收率较低。Jeremy等[15]在微波条件下,将芳香类含氟化合物在DMSO、叔丁醇钾和2-丁炔-1-醇的作用下合成芳香烃醚类,继而水解得到酚类,但其原料不易得。张晓薇[16]以Cu2O为催化剂,在高温高压条件下将间氯苯胺碱解制得间氨基苯酚,但能耗大、反应时间长。也有报道[17-19]以氯苯催化水解制备苯酚的方法,但对催化剂或工艺条件的要求较高。硼酸是一种高效且具有良好选择性的非金属催化剂,采用硼酸催化水解1,4-二氯蒽醌制备1,4-二羟基蒽醌既环保又经济。
1 实验部分
1.1 仪器与试剂
主要仪器:R-210型旋转蒸发仪、H1650型台式离心机、DF-101S型恒温加热磁力搅拌器、Agilent1200高效液相色谱仪、KQ-2200DE型数控超声波清洗器、NICOLET380型红外光谱仪、Unity INOVA300型核磁共振仪。
主要试剂:邻苯二甲酸酐、对二氯苯、五氯化磷、无水三氯化铝、无水氯化钙、硼酸、浓硫酸等,均为国产分析纯。
1.2 实验方法
1.2.1 1,4-二氯蒽醌的合成
取75.0g化合物1(图1)和115.0g PCl5,常温下加入到500mL斜型三口烧瓶中,加热至150℃反应12h,减压蒸馏除去POCl3,继续旋蒸收集131~133℃(1.2~1.3kPa)的棕黄色馏分,得到化合物2 (107.7g),收率为94.78%。取2(90%)45.0g和 25.0g对二氯苯加入到250mL的斜型三口烧瓶中,搅拌,加热至60℃预热。待对二氯苯完全溶解,将40.0g无水AlCl3和8.0g NaCl快速加入到烧瓶中,并立即升温至110~120℃,反应5~6h后将反应液倒入冰-水混合溶液中并迅速搅拌。静置分层,过滤,取滤饼放入200mL NaOH溶液(20%)中,沉淀水解,过滤,用36%浓盐酸调节pH值为1.5~2.0,得浅黑色沉淀粗体化合物3(45.2g),收率80.12%。3不需提纯处理,直接取5.0g 3加入到40.0g 98%浓硫酸中,150℃下反应4h后,倒入100mL水溶液中,过滤,得黄褐色沉淀[20]4(4.85g),产率89%。化合物4分析数据如下。1H NMR(500 MHz,CDCl3)δ:8.33~8.10 (m,2H,ArH),7.93~7.71 (m,2H,ArH),7.68 (s,2H,ArH);IR (KBr)ν/cm-1:3012,2653,2527,1690,1586,1497,1403,1282,1071,974,908,830,740,674,557。
1.2.2 1,4-二羟基蒽醌的合成
取1,4-二氯蒽醌5.0g、95%浓硫酸50.0g、硼酸3.0g,于220℃下共热反应60min,高效液相色谱法对反应过程进行中间控制分析。待1,4-二氯蒽醌峰消失后反应结束,过滤,得沉淀3.89g,用冰乙酸重结晶得粉红色结晶5。化合物5数据如下。1H NMR(500 MHz,CDCl3)δ:8.27 (dt,J = 24.0,12.1 Hz,2H,R-OH),7.87 (s,2H,ArH),7.88~7.56 (m,2H,ArH),5.26 (s,2H,ArH);IR (KBr)v/cm-1:3016,1630,1590,1452,1392,1330,1256,1148, 1148,1069,1027,965,875,836,792,726,699,580,498。
1.2.3 产物的纯度分析条件
1,4-二羟基蒽醌色谱条件,Agilent HC-C18 (250mm×4.6mm×5μm);流动相,V(甲醇)∶V(水)= 90∶10;流动相稀释样品,进样量20μL;流速1.0mL/min;柱温30℃;Agilent1200高效液相色谱仪。
2 结果与讨论
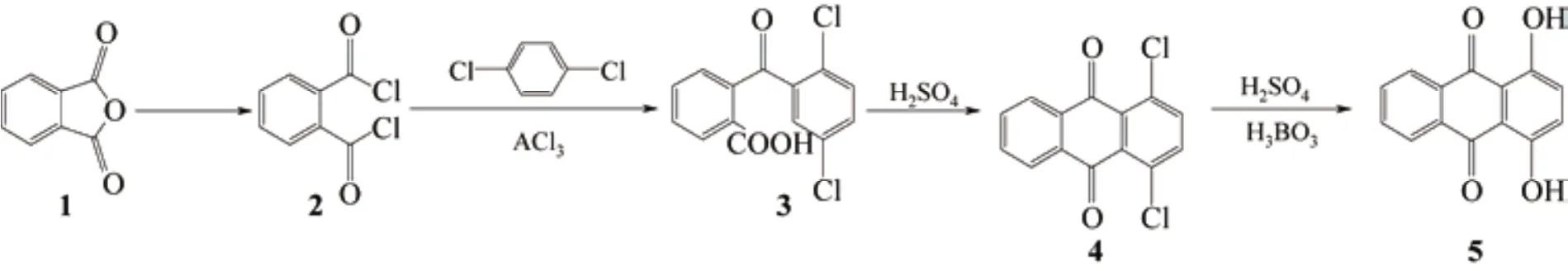
图1 1,4-二羟基蒽醌合成路线图
2.1 反应温度的影响
在硫酸质量分数95%、硫酸∶1,4-二氯蒽醌∶硼酸(摩尔比)为37.5∶1.0∶1.4、反应时间60min及反应温度200~240℃的条件下,考察温度对反应的影响,实验结果如图2所示。由图2可知,主产物的收率随温度的升高先增加后减小,且在220℃左右时达最大值,说明反应对温度敏感。温度过高,副反应加剧,因此,宜选择220℃作为反应温度。
2.2 硫酸质量分数的影响
在反应温度220℃、硫酸15mL、1,4-二氯蒽醌∶硼酸(摩尔比)为1.0∶1.4、反应时间60min及80%~105%硫酸的条件下,考察各浓度下硫酸介质对反应的影响,实验结果如图3所示。由图3可知,主产物的收率随着硫酸浓度的增加先增大后减小,在95%处达最大值,说明硫酸质量分数的增加有助于提高主产物收率。但当超过95%时收率开始下降,使用105%发烟硫酸时主产物收率降低得比较多,说明此时副反应加剧,反应倾向于发生磺化反应。因此,宜选择95%硫酸作为反应介质。
2.3 硼酸配比的影响
在反应温度220℃、95%硫酸∶1,4-二氯蒽醌(摩尔比)为37.5∶1.0、反应时间60min及硼酸加料量0.2~1.0g的条件下,考察硼酸的量对主产物收率的影响,实验结果如图4所示。由图4可知,随着硼酸的量的增加,产物的相对收率随之增加。但当加料量从0.6g增加到1.0g时,收率不再增加,说明此时催化已达到平衡。因此,选择加入硼酸的质量比为:硫酸∶1,4-二氯蒽醌∶硼酸=13.8∶1.0∶0.3,即原料中硫酸、1,4-二氯蒽醌、硼酸的摩尔比宜为37.5∶1.0∶1.4。
2.4 反应时间的影响
在反应温度220℃、硫酸∶1,4-二氯蒽醌∶硼酸(摩尔比)为37.5∶1.0∶1.4、95%硫酸的条件下,考察了反应时间对反应的影响。实验中注意到反应进行的比较剧烈,因此每隔10min对反应液取样进行高效液相色谱分析,得到主产物峰面积及峰面积比(监测得到的产物峰面积/1,4-二羟基蒽醌标样峰面积)的变化趋势如图5所示。由图5可知,在实验考察的范围内,反应前60min内主产物峰面积变化曲线不断上升,峰面积比曲线也跟着上升,说明主产物的量在不断增加。而60min之后,两条曲线略有下降,说明主产物的量在减少。因此,反应时间应维持在60min。
3 反应机理讨论

图2 反应温度的影响
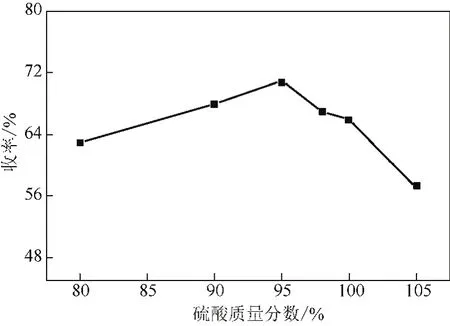
图3 硫酸浓度的影响
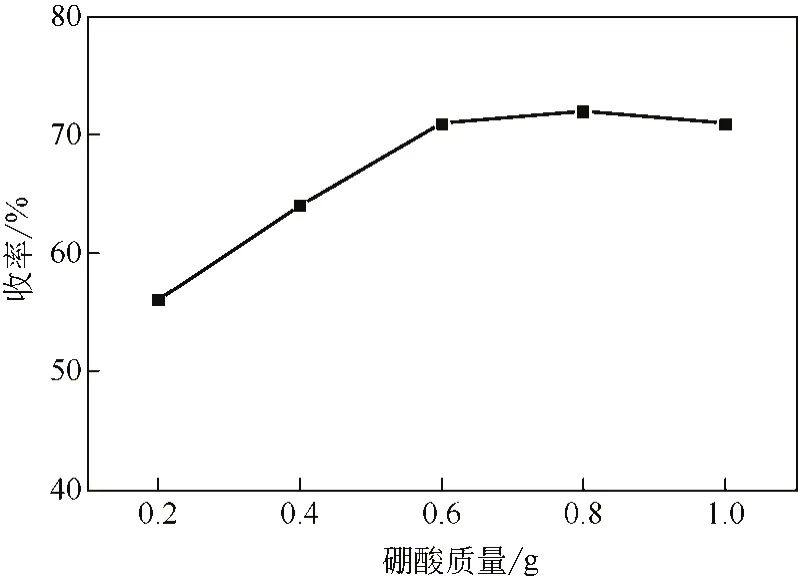
图4 硼酸加料量影响
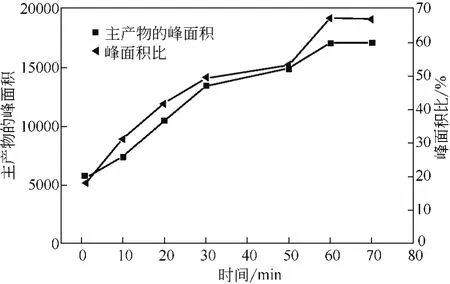
图5 反应时间的影响
化合物4水解得到化合物5的反应主要是蒽醌结构中1、4位上基团的取代反应,即发生在蒽醌的苯环结构上的取代反应[21]。因此,1、4位上的基团发生取代反应的难易程度取决于苯环上官能团对苯环的活化程度。苯环上的取代是亲电取代反应[22]。从反应活性的角度看,凡有助于提高苯环上电子云密度的基团,就能使苯环活化,反应活性提高,反之则使苯环钝化,降低反应活性;从反应位置的角度看,若苯环上有取代基,则取代基的电子效应沿着苯环共轭体系传递,即当与苯环上相连的原子含有未共用电子对取代基团时,电子间的相互作用使未共用电子对(简称p电子)与苯环形成p-π共轭体系,从而使得整个苯环具有给电子效应,常见的给电子基团如—NR2、—NHR、—NH2、—OH、—OR、—X等[23]。因此,—NH2、—OH等基团会使苯环活化,而对卤素元素而言,由于其较强的吸电诱导效应,使得苯环上的电子云密度降低,对苯环有钝化作用。由此可知,由于化合物4含有两个氯原子,苯环活性较低,使得反应条件苛刻,反应需在催化剂的作用下进行。据此,研究并提出了化合物4水解得到化合物5的可能的反应机理(见图6):一分子硼酸在H2O作用下,形成,亲核进攻苯环羰基C,通过双键电子传递,活化苯环,在酸性环境下水解脱除Cl-,再结合一分子H2O得到化合物10。同理,另一分子硼酸再结合对位的羰基与羟基,最终水解得到化合物5。

图6 化合物4水解得到化合物5可能的反应机理
4 结 论
设计了一条合成1,4-二羟基蒽醌的路线,并探索和解释了1,4-二氯蒽醌水解得到1,4-二羟基蒽醌可能的机理。通过单因素试验的方法得到氯转羟基反应较优的反应条件:反应温度为220℃、硫酸浓度为95%,硫酸∶1,4-二氯蒽醌∶硼酸(摩尔比)为37.5∶1.0∶1.4,反应时间为60min。在此条件下经重复试验得到1,4-二羟基蒽醌的收率为71.0%。该方法原辅料廉价易得,高效简易,为1,4-二羟基蒽醌的工业生产提供了一种新的方法参考。
[1] 陆阳. 醌类化学[M]. 北京:化学工业出版社,2009:65-67.
[2] 殷卫峰,欧植泽,高云燕. 1,4-二羟基蒽醌及其Y3+配位聚合物与DNA相互作用研究[J]. 化学学报,2010,68(14):6.
[3] 杨希川,吴祖望. 蒽醌型酸性、分散和活性染料及其中间体生产技术的进展[J]. 化工进展,2002,21(6):386-390.
[4] 柯贵珍,于伟东,徐卫林. 药用植物染料的特征和功能实(I):药性、颜色与染色 [J]. 武汉科技学院学报,2006,19(1):44.
[5] 李春梅. 1,4-二羟蒽醌是一种有效的重组人蛋白激酶CK2的抑制剂[J]. 齐齐哈尔医学院学报,2011,32(14):2.
[6] 宋晓凯. 天然药物化学[M]. 北京:化学工业出版社,2010:59-60.
[7] 李坤兰,刘珊珊. 纺织用1,4-二羟基蒽醌染料的合成[J]. 大连工业大学学报,2009,28(4):1-2.
[8] 赵欣. 葸醌类靶向抗肿瘤化合物的合成与性质研究[D]. 天津:天津理工大学,2012.
[9] 谷晓丽. 1,4-二羟基蒽醌的合成研究[J]. 辽宁化工,2000,29(3):131-132.
[10] 康晓丽,欧阳福生. 苯酐法合成蒽醌固体酸催化剂的进展[J]. 精细化工中间体,2005,35(4):4-7.
[11] Breslin D T,Coury J E,Anderson J R,et al. Anthraquinone photonuclease structure determines its mode of binding to DNA and the cleavage chemistry observed[J].Journal of the American Chemical Society,1997,119(21):5043-5044.
[12] El’tsov A V,Sokolova N B,Grigor’ev A D,et al. Synthesis of quinizarin and its derivatives under conditions of microwaveactivation[J].Russian Journal of General Chemistry,2002,72(2):255-258.
[13] Krapcho Paul A,Waterhouse D. Sodium trimethyl silanoate. A hydroxyl synthon for fluoride SNAr type displacements from anthracene-9,10-dione,benz[g] isoquinoline-5-10-diones and Nitrobenzenes[J].Synthetic Communications,1998,28(18):3415-3422.
[14] 杨慧慧,张跃,严生虎,等. 间二氯苯水解制备间苯二酚[J].南京工业大学学报,2007,29(2):30-33.
[15] Jeremy I L,Mila T Du. Rapid,one-pot conversion of aryl fluorides into phenols with 2-butyn-1-ol and potassiumt-butoxide in DMSO[J].Synthetic Communications,2002,32(9):1401-1406.
[16] 张晓薇. 间氯苯胺加压碱解法制备间氨基苯酚工艺研究[J]. 上海化工,2012,32(9):11-13.
[17] 陈灵晶,林海强,杨乐夫. 固态离子交换法合成Cu/HZSM-5催化剂及其氯苯气相羟基化制苯酚催化性能的研究[C]//第十三届全国催化学术会议论文集,兰州,2006.
[18] 陈培丰,陈守正. 氯苯气相水解制苯酚的离子交换负载型磷酸盐催化剂的研究[J]. 福建师范大学学报,1993,9(1):41-45.
[19] 黄颖,童跃进,陈守正. TiAPO-5分子筛负载磷酸镧铜催化氯苯气相水解制苯酚[J]. 福建化工,2000(1):18-21.
[20] 杨新颖. 1,4-二氯蒽醌的制备[J]. 中国医药工业杂志,2007,38 (4):266-281.
[21] Joseph P A,Priyadarshini S,Kantam M L,et al. Sulfonic acid resin and copper salts:A novel heterogeneous catalytic system for direct hydroxylation of haloarenes[J].Catalysis Science & Technology,2011,1(4):582-585.
[22] 高鸿宾. 有机化学[M]. 北京:高等教育出版社,2005:169-179.
[23] 周子英. 取代基对苯环活性的影响[J]. 内蒙古石油化工,2003,29(2):18-19.
Preparation of 1,4-dihydroxyanthraquinone from hydrolysis of 1,4-dichloroanthraquinone
WANG Guicheng,DUAN Zhengkang,YAN Zhixiang
(School of Chemical Engineering,Xiangtan University,Xiangtan 411105,Hunan,China)
:1,4-dihydroxyanthraquinone was obtained by boric acid catalytic hydrolysis of 1,4-dichloroanthraquinone,which was synthesized by using commercial phthalic anhydride andp-dichlorobenzene as raw materials. The effect of reaction temperature,sulfuric acid concentration (mass concentration),molar ratio of reactants and reaction time on the chlorine hydroxylation reaction was investigated,and optimal experimental conditions were obtained as follows,temperature 220℃,sulfuric acid concentration 95%,molar ratio of sulfuric acid/1,4-dichloroanthraquinone/boric acid 37.5∶1.0∶1.4,reaction time 60min. Under above conditions,the yield of 1,4-dihydroxyanthraquinone was 71.0%. The possible mechanism of chlorine hydroxylation with the catalytic effect of boric acid was discussed.
1,4-dihydroxyanthraquinone;synthesis;chlorine hydroxylation;mechanism
TQ 612.5
A
1000-6613(2014)11-3053-04
10.3969/j.issn.1000-6613.2014.11.036
2014-02-26;修改稿日期:2014-03-20。
王贵城(1988—),男,硕士研究生,主要从事精细化学品合成与分析联系人:段正康,教授。E-mail dzk0607@163.com。
