核壳结构碳化钨/碳化钨铁复合材料的制备与电催化活性
2014-06-23谢伟淼李国华
陈 辉 陈 丹 谢伟淼 郑 翔 李国华,2,3,*
(1浙江工业大学化学工程学院,杭州310032;2绿色合成技术国家重点实验室培育基地,杭州310032;3浙江工业大学纳米科学与工程技术研究中心,杭州310032)
1 引言
碳化钨因具有类铂催化性能,1,2可用作化学催化和电催化领域的催化剂,3-5且在催化反应过程中不易被CO、H2S等气体中毒6而受到了广泛关注.碳化钨的上述特性使其具备了替代或部分替代铂等贵金属催化剂的潜质.现有研究表明,碳化钨的催化活性远不及铂等贵金属.7,8因此,如何提高碳化钨催化活性,并使其接近铂等贵金属催化剂是碳化钨替代和部分替代铂等贵金属催化剂的关键,也是碳化钨基催化材料研究的热点和走向工业应用的核心问题之一.
金属化的表面结构是碳化钨具有类铂催化性能的原因所在,9若能够调控碳化钨的表面电子结构,必可调控碳化钨的催化活性.基于这样的认识,国内外专家学者开展了不少有针对性的研究工作,10-19比如,采用模板法将碳化钨制备成具有一定结构和形貌催化材料,10,11应用原位还原碳化和碳包覆将碳化钨制备成纳米材料,12-18将碳化钨与其它材料复合,19这些均不同程度地提高了碳化钨的催化活性.本课题组在这方面也开展了一系列研究工作.首先,利用碳纳米管优越的电子性能、管状结构和比表面积大等特点,将偏钨酸铵负载于硝化后的碳纳米管上,经还原碳化后获得了碳化钨与碳纳米管复合材料;20其次,以具有三维联通孔道结构的天然沸石为载体制备了碳化钨与天然沸石纳米复合材料;21再次,利用Ti4+的空d轨道的独特性质,22以金红石型纳米二氧化钛为载体制备了碳化钨与金红石纳米复合材料;23最后,以铁黄为载体,偏钨酸铵为钨源,将表面包覆法与原位还原碳化技术相结合,制备出了具有核壳结构的碳化钨与碳化二钨纳米复合材料.24上述复合材料均在一定程度上提高了碳化钨的电催化活性,但也面临一些问题.碳纳米管虽具有优越的电子性能和管状结构,但在电化学环境下不稳定会被氧化,从而导致复合材料的结构与性能发生变化.以天然沸石和二氧化钛为载体虽然可克服上述问题,但其电催化活性和导电性较差,且复合材料中碳化钨分布的均匀性也较难控制.
为寻找解决上述问题的技术方法,本文提出制备碳化钨铁(Fe3W3C)与碳化钨(WC)核壳结构复合材料的新思路.这是因为,一方面,Fe3W3C和WC均对甲醇的电催化氧化具有较好的催化活性;25另一方面,Fe3W3C具有良好的导电性;更为重要的是核壳结构复合材料可利用核和壳层的特点对材料的性能进行调控,可明显提升复合材料的催化性能.26基于上述因素,本文利用铁黄比表面积大和吸附性能优越等特点,采用包覆技术将偏钨酸铵均匀地吸附于其外表面,并利用铁黄中的铁元素在还原碳化过程中能够掺入碳化钨内形成Fe3W3C,以增强复合材料导电性的特点,将表面包覆和原位还原碳化技术相结合,制备出具有核壳结构的Fe3W3C与WC复合材料,并首次报道了Fe3W3C与WC核壳结构复合材料对甲醇的电催化性能.
2 实验部分
2.1 样品制备
前驱体制备.称取一定量的偏钨酸铵(AMT,分析纯,湖南株洲硬质合金厂),溶解于400 mL去离子水中,再按比例称取相应质量的铁黄(FeOOH,分析纯,浙江升华集团德清华源颜料有限公司)加入其中,在恒温磁力搅拌器内60°C条件下持续搅拌3 h,静置,去上层清液后于烘箱内80°C干燥10 h,研磨即得前驱体.
样品制备.将前驱体装于石英舟内,再置于恒温管式炉中,以H2为还原气体、CH4为碳源,在900°C条件下还原碳化4 h,冷却后即得碳化钨铁与碳化钨复合材料.
系列样品制备.按上述方法分别制备AMT与FeOOH质量比5、10、15和20的四个样品,并分别标注为样品1、2、3和4.
2.2 样品表征
样品物相,即X射线衍射分析(XRD)分析采用荷兰PANalytical公司生产的X′Pert PRO型X射线衍射仪,测试时采用CuKα射线源(λ=0.1541 nm),工作电压为40 kV,电流为40 mA,扫描范围5°-80°,记数步长0.033°.XRD数据用仪器自带分析软件High Score进行处理,处理过程中仪器本征宽化采用厂家提供的多晶硅标准样品进行校正与扣除;样品形貌采用场发射扫描电子显微镜(SEM Hitachi S-4700II)进行观察;样品微结构采用高分辨透射电子显微镜(TEM Tecnai G2 F30)进行表征;样品的化学组成采用X射线能量散射谱(EDS)进行分析.
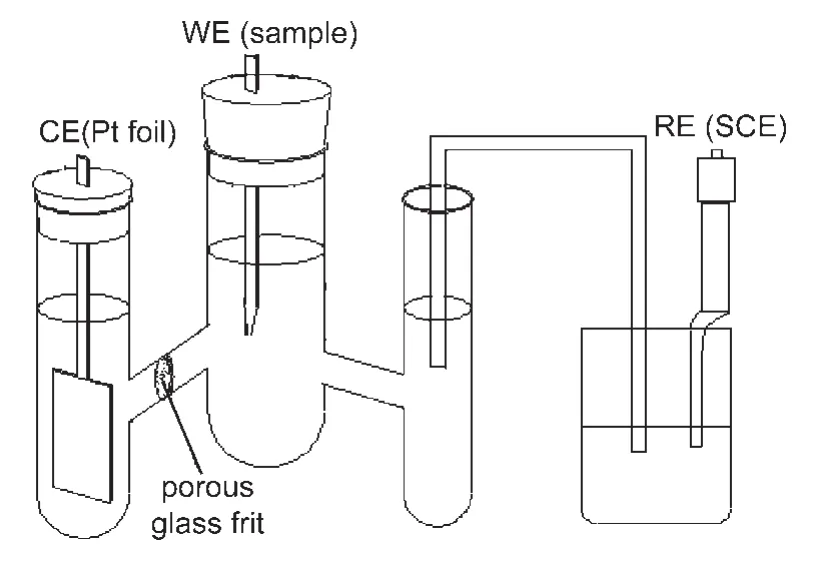
图1 三电极体系示意图Fig.1 Schematic diagram of tri-electrode system
2.3 电化学性能测试
样品的电催化性能用CHI620B电化学工作站测试.测试过程中采用三电极体系(如图1所示)、测试时用循环伏安法(CV),工作电极(WE)为粉末微电极,27参比电极(RE)为饱和甘汞电极(SCE),辅助电极(CE)为光亮铂电极,盐桥由氯化钾与琼脂质量比为10:1的混合物制备而成.测试前为了形成稳定的三相反应界面,将电极在电解液中浸泡3 h.测试过程中溶液电阻采用正反馈技术进行欧姆补偿.
3 样品表征
3.1 前驱体样品表征
3.1.1 XRD表征
图2为前驱体的XRD结果.从图2中可以看到,前驱体的XRD图中有十多处衍射峰,其中,2θ为8.5560°、9.9328°、27.8638°等处的衍射峰可归属为钨酸铵(PDF No.070-2271),2θ为 21.2159°、33.2233°、36.6426°等处的衍射峰可归属为FeOOH(PDF No.081-0463),即前驱体主要含有两种物相:钨酸铵(APT)和FeOOH.这说明,将偏钨酸铵溶于水并负载于铁黄表面之后,铁黄的物相基本没有改变,偏钨酸铵转变成了钨酸铵.

图2 前驱体的XRD图谱Fig.2 X-ray diffraction(XRD)pattern of the precursor APT:ammonium tungstate
3.1.2 SEM表征
图3是前驱体的扫描电子显微镜(SEM)照片,其中,图3A为前驱体的形貌,图3B为前驱体的表面结构.从图3B中可看出,前驱体大多呈短小柱状,长短不一,柱体的平均长度在500 nm左右,宽80-150 nm,柱体表面比较光滑.这与铁黄载体的形貌24有一定的差异.这是由于在前驱体制备过程中,经过搅拌、加热、干燥以及研磨后,载体铁黄的形貌有一定程度的破坏,负载后的前驱体在干燥和研磨过程中也会断裂.
3.1.3 TEM表征
图4为前驱体的透射电镜(TEM)照片.24从图4A中可看出,前驱体呈棒状,保持了铁黄的形貌,长度多在500 nm以上,粗细不等,直径50-70 nm;与铁黄(见图4A中嵌入图)相比,前驱体中间灰度较高,边缘灰度较低,两者之间有明显的界线.这反映前驱体可分为载体和负载层两个部分,后者厚度6.5-9.0 nm,如图4A所示.图4B为的图4A的局部放大,经测量,负载层的晶面间距为0.7858 nm,与APT(PDF No.070-2271)的(111)晶面间距接近.这与XRD的分析结果一致.依据上述结果可认为,前驱体是由APT和FeOOH组成,且负载层为APT,载体为FeOOH,两者构成了具有典型核壳结构的棒状前驱体.

图3 前驱体的SEM照片Fig.3 Scanning electron microscope(SEM)images of the precursor

图4 前驱体的透射电子显微镜照片24Fig.4 Transmission electron microscope(TEM)images of precursor24
3.2 样品表征
3.2.1 XRD表征
图5是以偏钨酸铵和铁黄为原料,按不同AMT与FeOOH比例制备的前驱体经还原碳化后样品的XRD分析结果.从图5中可看到,随着制备过程中AMT与FeOOH比例的变化,样品的主要衍射峰没有明显变化,其中,2θ为 13.79°、22.64°、32.25°、35.24°、39.78°、42.31°、46.26°、59.48°、64.49°、69.30°、72.23°和73.99°处的衍射峰可归属于碳化钨铁 Fe3W3C(PDF No.94-004-3230),且 2θ为 42.31°、39.78°、32.25°、35.24°和46.26°处的衍射峰是碳化钨铁的特征峰;2θ为35.56°处的衍射峰可归属于碳化钨WC(PDF No.051-0939)的特征峰,即最强峰;2θ为38.13°处的衍射峰可归属于碳化二钨W2C(PDF No.089-2731)的特征峰.这说明四个样品的主要物相均为Fe3W3C、WC和W2C.其中,后两种物相只看到了最强衍峰,其它衍射峰未能看到的原因是它们被碳化钨铁的衍射峰掩盖所致.
比较上述四个样品WC和W2C的衍射峰强可知,样品1中WC的特征峰衍射强度较弱,W2C特征峰衍射强度较强;与样品1相比,样品2中WC特征峰的衍射强度明显增强,W2C特征峰的衍射强度却有所下降;与样品2相比,样品3中W2C特征峰的衍射强度没有较大改变,但WC特征峰的衍射强度增强;样品4中WC和W2C特征峰的衍射强度均较弱.上述结果说明,W2C特征峰的衍射强度随着制备过程中AMT与FeOOH比例的增加而持续减弱;WC特征峰的衍射强度在AMT与FeOOH比例为5-15范围内持续增强,当这一比例为20时却明显减弱.对于同一物相而言,衍射峰强度与物质的量有着一定的相关性.据此,上述结果说明在样品制备过程中,当AMT与FeOOH的比例在5-20范围内时,随着比例的增大,W2C含量逐渐降低;当比例在5-15范围内时,WC含量随着比例的增大而逐渐增加,当比例增大到20时,WC含量下降.

图5 AMT与FeOOH不同质量比样品的XRD图谱Fig.5 XRD patterns of the samples prepared with different mass ratios ofAMT to FeOOH
比较样品中F3W3C的衍射峰强度可知,样品4的衍射峰强度最高,其次是样品2,再是样品3,最弱的是样品1.这说明样品4中F3W3C的含量最多,其次是样品2,再是样品3,含F3W3C最少的是样品1.
3.2.2 SEM
图6为样品2的扫描电子显微镜照片.从图6中可看出,样品的颗粒为不规则粒状,粒径介于150-500 nm之间,颗粒粒径较为均匀,分散相对较好.这与其前驱体的棒状形貌有明显差别.这是因为前驱体在高温还原碳化过程中发生了融熔和重结晶,从而导致样品颗粒的形貌由棒状转变成了颗粒状.
3.2.3 TEM
图7为样品的TEM照片,其中A、B、C和D分别为样品1、2、3和4的整体形貌.从图7中可以看到,样品1、2和3的颗粒粒径均在200-500 nm范围内,样品4的颗粒粒径下降到200 nm以下,即随着制备过程中AMT与FeOOH比例的增加样品的颗粒粒径逐渐减小.图7中的四个嵌入图均为各自样品颗粒的局部放大.从图7中可看出,样品1中颗粒的核壳结构不是很清晰,如图7A中嵌入图所示,其余3个样品颗粒的核壳结构清晰可见,壳层的厚度均在100 nm以下,如图7B、7C和7D中嵌入图所示.比较核壳结构的壳层厚度和均匀性,由样品2到样品4,壳层的厚度逐渐减小,均匀性逐渐增加.这说明样品的微结构和AMT与FeOOH比例相关.

图6 样品2的扫描电子显微镜照片Fig.6 Scanning electron microscopy(SEM)image of the sample 2

图7 样品的TEM照片Fig.7 TEM images of the samples A:sample 1;B:sample 2;C:sample 3;D:sample 4
为了解样品颗粒的核部与壳层的晶相组成,测量了样品2中壳层和核部的晶面间距,结果如图7B中所示,其中,壳层中有一组清晰的晶格条纹,其间距为0.2451 nm,这与碳化钨(PDF No.051-0939)的(100)晶面间距接近,说明壳层以碳化钨为主;核部有一组清晰的晶格条纹,其间距为0.6578 nm,这与碳化钨铁(PDF No.089-2579)的(111)晶面的晶面间距接近,说明核部以碳化钨铁为主.上述结果说明样品2的颗粒具有以碳化钨铁为核,碳化钨为壳的核壳结构.由于样品3和4的制备方法与样品2相同,且XRD分析结果表明三者的晶相组成相同.据此可认为,样品2、3和4均具有以碳化钨铁为核,碳化钨为壳的良好核壳结构.
3.2.4 EDS表征
图8是样品的EDS分析结果.从图8中可以看出,样品主要含有C、Fe、W和Cu元素,其中,Cu来自测试使用的铜网,C部分来自测试使用的碳膜,部分来自样品,即样品的主要成份为C、Fe和W.
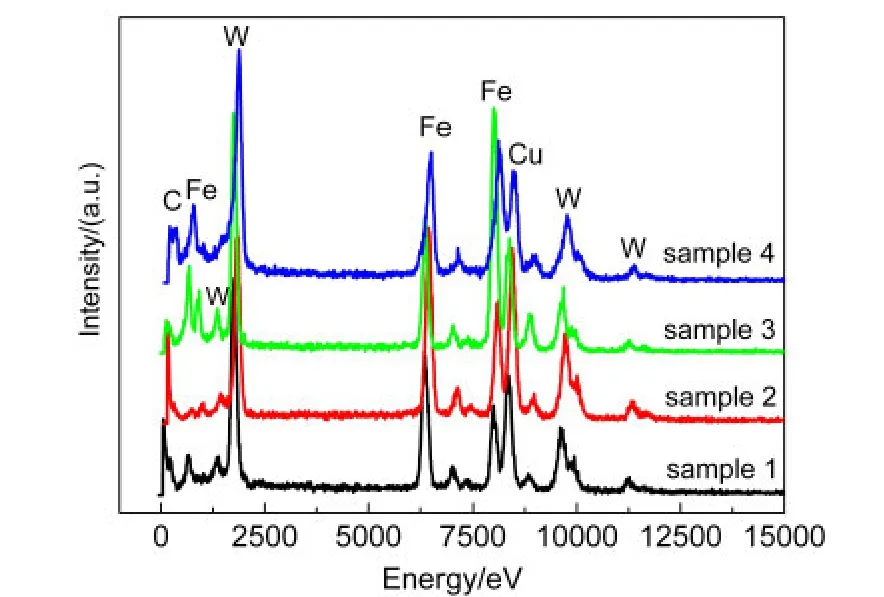
图8 AMT与FeOOH不同质量比样品的EDS图谱Fig.8 Energy dispersive spectroscopy(EDS)patterns of the samples prepared with different mass ratios ofAMT to FeOOH
综上所述,结合样品的XRD、TEM和EDS分析结果,可以推断:样品呈典型的核壳结构,核部主要物相为Fe3W3C,壳层主要物相为WC和W2C.
3.3 电化学性能
3.3.1 碱性体系
图9是样品1、2、3和4,以及介孔空心球状碳化钨(meso WC)和颗粒状碳化钨(part WC)在0.25 mol·L-1CH3OH+0.1 mol·L-1NaOH溶液中的循环伏安曲线.从图9中可以看到,六个样品的扫描曲线均出现了一个明显的氧化峰和一个还原峰.为便于对比,将图9中各样品氧化峰的峰电位和峰电流列于表1.
比较各样品的氧化峰电位值可知,峰电位从低到高依次为:样品1<样品3<样品4<样品2<meso WC<part WC.与meso WC的氧化峰电位相比,样品1、3、4和2的氧化峰电位分别负移0.3200、0.3100、0.2867和0.1749 V.这说明样品在碱性溶液中对CH3OH的电催化氧化性能均优于meso WC和part WC,且样品1的催化性能最佳,样品2的催化性能最差.

图9 样品在0.25 mol·L-1CH3OH+0.1 mol·L-1NaOH溶液中的循环伏安曲线Fig.9 CV curves of the samples in 0.25 mol·L-1 CH3OH+0.1 mol·L-1NaOH solution
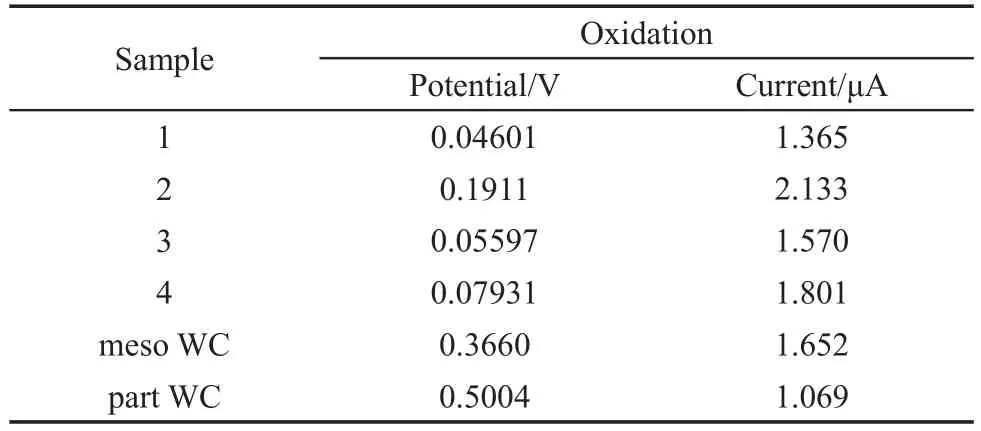
表1 图9中的氧化峰电位与及电流Table 1 Potentials and currents from Fig.9
从表1中的数据可知,电流从大到小依次为:样品2>样品4>meso WC>样品3>样品1>part WC.这说明在碱性溶液中样品2的导电性最佳,所有样品的导电性均优于part WC,且样品2和样品4的导电性还优于meso WC.
3.3.2 中性体系
图10是样品1、2、3和4,以及meso WC和part WC在 0.25 mol·L-1CH3OH+0.1 mol·L-1NaCl溶液中的循环伏安曲线.从图10中可以看到,六个样品的扫描曲线均出现了一个氧化峰,除了样品1外,其余样品的扫描曲线均出现了还原峰.为了便于比较样品的电催化性能,将样品的氧化峰电位和电流列于表2.
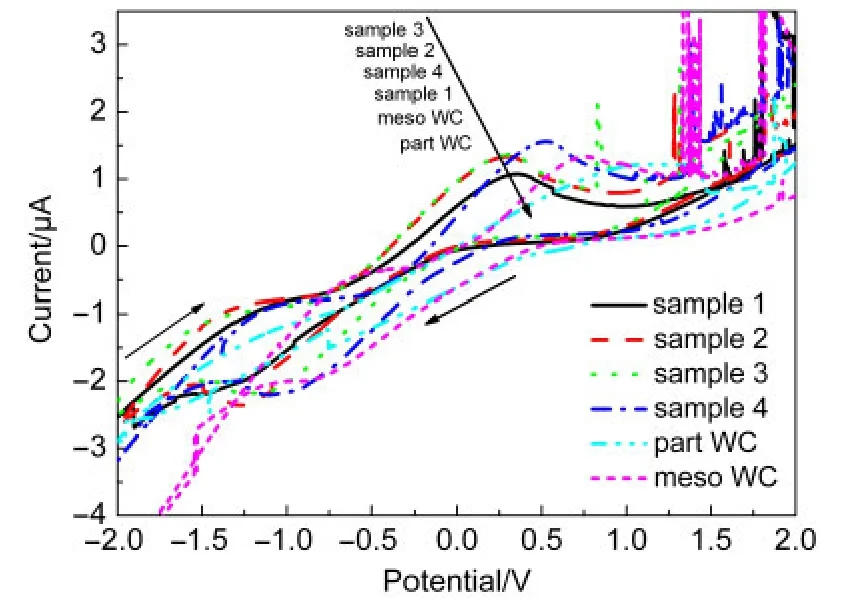
图10 样品在0.25 mol·L-1CH3OH+0.1 mol·L-1 NaCl溶液中的循环伏安曲线Fig.10 CV curves of the samples in 0.25 mol·L-1 CH3OH+0.1 mol·L-1NaCl solution
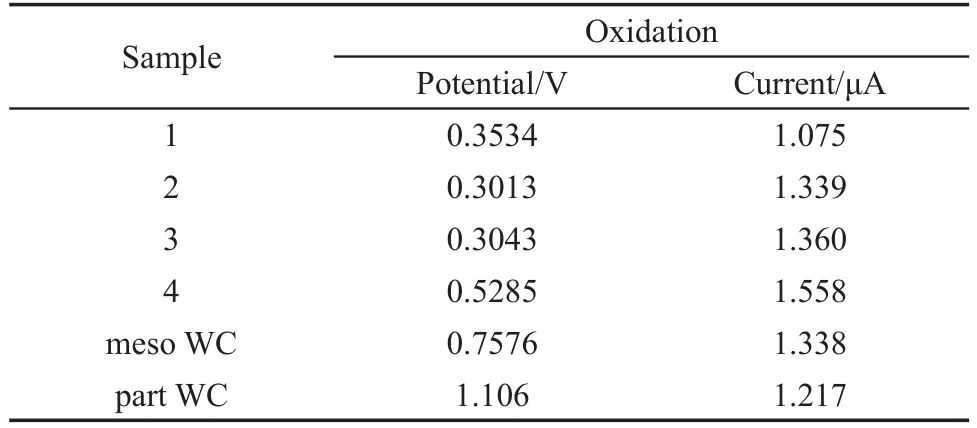
表2 图10中氧化峰电位及电流Table 2 Potentials and currents from Fig.10
从表2中的数据可知,样品的氧化峰电位从低到高依次为:样品2<样品3<样品1<样品4<meso WC<part WC.与meso WC的氧化峰电位相比,样品2、3、1和4的氧化峰电位分别负移0.4563、0.4543、0.4042和0.2291 V.这说明在中性溶液中,对CH3OH的电催化氧化性能最佳的为样品2,其次是样品3和1,最差是样品4,且均优于meso WC和part WC.
比较各样品的氧化峰电流可知,电流从大到小依次为:样品4>样品3>样品2>meso WC> part WC>样品1.这说明在中性溶液中,除样品1外,其余样品的导电性均优于meso WC和part WC,导电性最佳的是样品4.
3.3.3 酸性体系
图11是样品1、2、3和4,以及meso WC和part WC在0.25 mol·L-1CH3OH+0.1 mol·L-1HCl溶液中的循环伏安曲线.从图11中可以看到,六个样品的扫描曲线均出现了一个氧化峰和一个还原峰.为了便于比较样品的电催化性能,将样品的氧化峰电位和电流列于表3.

图11 样品在0.25 mol·L-1CH3OH+0.1 mol·L-1 HCl溶液中的循环伏安曲线Fig.11 CV curves of the samples in 0.25 mol·L-1 CH3OH+0.1 mol·L-1HCl solution

表3 图11中的电位及电流Table 3 Potentials and currents from Fig.11
从表3中的数据可知,样品氧化峰电位从低到高依次为:样品1<样品2<样品3<样品4<meso WC<part WC.与meso WC的氧化峰电位相比,样品1、2、3和4的氧化峰电位较分别负移0.4978、0.4472、0.3635和0.1232 V.这说明样品在酸性溶液中对CH3OH的电催化氧化性能均优于meso WC和part WC.
比较样品的氧化峰电流值可知,电流从大到小依次为:样品4>样品3>样品1>样品2>meso WC>part WC.这说明样品在酸性溶液中的导电性也优于meso WC和part WC,且导电性最佳的是样品4.
4 分析与讨论
4.1 溶液性质对性能的影响
图12是样品1在碱性、中性和酸性溶液中的循环伏安曲线.从图12中可以明显观察到,样品1扫描曲线的氧化峰电位从低到高依次为:碱性<中性<酸性.这说明样品1在碱性溶液中对CH3OH的电催化氧化性能最佳,其次是在中性溶液中,最差是在酸性溶液中.
这是因为甲醇的电催化氧化反应,28在酸性溶液中的总反应式为:

在碱性溶液中的总反应式为:

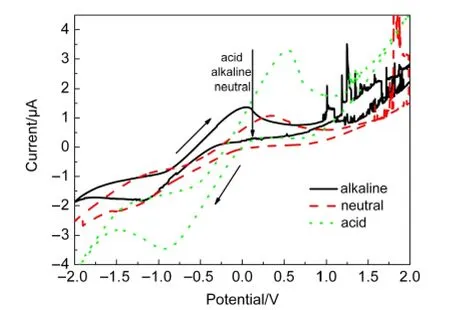
图12 样品1在各种CH3OH溶液中的循环伏安曲线Fig.12 CVcurves of the samples in different CH3OH solutions
由此可知,甲醇在酸性和碱性溶液中的电催化反应有所不同,酸性溶液中H+主要起导电作用,而不参与实际的反应;碱性溶液中OH-不仅具有导电作用,还参与具体的化学反应,即反应体系中OH-的量越多的,越能促进反应向右进行.
比较样品1在不同溶液中的导电性,发现其在酸性溶液中的氧化峰电流明显高于在碱性和中性溶液中的电流.这是与酸性溶液中H+浓度较高且参与导电有关.
将样品1、2、3和4,以及meso WC和part WC对CH3OH的电催化氧化性能进行比较发现,在碱性溶液中,样品对CH3OH的电催化性能从强到弱依次为:样品1>样品3>样品4>样品2,且均优于meso WC和part WC;在中性溶液中,样品对CH3OH的电催化性能从强到弱依次为:样品2>样品3>样品1>样品4,且均优于meso WC和part WC样品;在酸性溶液中,样品对CH3OH的电催化氧化性能从强到弱依次为:样品1>样品2>样品3>样品4,且均优于meso WC和part WC样品.比较样品在不同性质溶液中的氧化峰电位高低可发现,样品在碱性溶液中的氧化峰电位最低,其次是在中性溶液中,最差是在酸性溶液中,即样品在碱性溶液中的电催化活性最高,其次是在中性溶液中,最差是在酸性溶液中.这说明样品在碱性溶液中的催化性能均优于其在中性和酸性溶液中的催化性能.这是因为CH3OH的电催化氧化反应须有大量OH-的参与,碱性CH3OH溶液中有充足的OH-,从而促进CH3OH氧化反应的进行,致使样品在碱性溶液中的电催化氧化性能优于其在中性和酸性溶液中的催化性能.
4.2 物相与性能关联性
从图9-12中可看出,当电势电位高于1.0 V时,在不同的体系中均出现了明显的尖锐峰.这是由于样品中含有少量铁,以及碳化钨铁中的铁在循环伏安过程中能与溶液中的H+离子发生电子交换反应,并释放出一定量的氢气所致.
由于中性溶液中OH-浓度与H+的浓度处于平衡状态,因此,在中性溶液中的催化性最能体现样品的本征特性.样品本征催化活性的强弱顺序为样品2>样品3>样品1>样品4.依据XRD分析结果,样品1中W2C和WC的含量最多,其次是样品2和样品3,样品4中WC和W2C的含量最少.据此可以推断,样品中对CH3OH起主要催化作用的是WC和W2C.
中性溶液中不同样品的导电性能为:样品4>样品3>样品2>样品1.结合样品的XRD分析结果,四个样品中样品4的Fe3W3C含量最高,样品1的Fe3W3C含量最低.这说明样品中Fe3W3C一方面作为载体,另一方面主要起导电作用.
综上所述,无论在碱性、中性还是酸性溶液中,碳化钨/碳化钨铁复合材料对CH3OH的电催化氧化性能均优于meso WC和part WC;碳化钨/碳化钨铁复合材料中在不同溶液中的催化性能具有一定的规律性,并与样品的物相组成有一定的相关性,对于CH3OH的电催氧化反应而言,样品的催化活性主要与WC和W2C的含量相关,样品的导电性主要与Fe3W3C的含量相关.
5 结论
以偏钨酸铵为钨源,以铁黄为铁源,将表面包覆与原位还原碳化技术结合可成功制备具有核壳结构的碳化钨铁和碳化钨复合材料;核壳结构的特征是以碳化钨铁为核,以碳化钨和碳化二钨为壳.
核壳结构复合材料在碱性、中性和酸性溶液中对甲醇均具有一定的催化活性,且均高于介孔空心球状碳化钨和颗粒状碳化钨的电催化活性;在上述三种体系中,以碱性溶液中的电催化活性最高,其次是中性溶液,酸性溶液最差;核壳结构复合材料的电催化活性不仅与其物相组成有关,还与其微结构特征相关.这些结果说明,复合材料的电催化活性可以通过改变反应体系的性质,以及控制材料本身的物相和结构特征进行调控.上述结果充分说明核壳结构是提高碳化钨电催化活性的有效途径之一.
(1) Levy,R.B.;Stauffer,M.C.Science1973,181,547.doi:10.1126/science.181.4099.547
(2) Böhm,H.Nature1970,227,484.
(3)Wang,G.J.;Liu,R.Z.;Chang,J.S.J.Qingdao Univ.2001,16(3),51 [王广建,柳荣展,常俊石.青岛大学学报,2001,16(3),51.]
(4) Zhu,L.Z.;Chen,Y.F.;Zhang,Q.Y.Chin.J.Appl.Chem.1999,16(4),52.[朱龙章,陈宇飞,张庆元.应用化学,1999,16(4),52.]
(5)Ma,C.A.;Yang,Z.W.;Zhou,Y.H.;Zha,Q.X.Acta Phys.-Chim.Sin.1990,6(5),622.[马淳安,杨祖望,周运鸿,查全性.物理化学学报,1990,6(5),622.]doi:10.3866/PKU.WHXB19900521
(6) Palanker,V.S.;Gajyev,R.A.;Sokolsky,D.V.Electrochim.Acta1977,22,133.
(7) Zellner,M.B.;Chen,J.G.Catal.Today2005,99,299.doi:10.1016/j.cattod.2004.10.004
(8) McIntyre,D.R.;Burstein,G.T.;Vossen,A.J.Power Sources2002,107,67.doi:10.1016/S0378-7753(01)00987-9
(9) Günter,S.E.;Detlef,B.;Walter,S.J.Catal.1976,43,353.doi:10.1016/0021-9517(76)90321-3
(10)Chen,Z.Y.;Shi,M.Q.;Ma,C.A.;Chu,Y.Q.;Zhu,A.J.Power Technology2013,235,467.doi:10.1016/j.powtec.2012.10.013
(11)Bosco,J.P.;Sasaki,K.;Sadakane,M.;Ueda,W.;Chen,J.G.Chem.Mater.2010,22,966.doi:10.1021/cm901855y
(12)Cui,X.Z.;Zhou,X.X.;Chen,H.R.;Hua,Z.L.;Wu,H.X.;He,Q.J.;Zhang,L.X.;Shi,J.L.International Journal of Hydrogen Energy2011,36,10513.doi:10.1016/j.ijhydene.2011.06.050
(13)Yan,Y.;Zhang,L.;Qi,X.;Song,H.;Wang,J.Y.;Zhang,H.;Wang,X.Small2012,21,3350.
(14) Giordano,C.;Antonietti,M.Nano Today2011,6,366.doi:10.1016/j.nantod.2011.06.002
(15) Shen,P.K.;Yin,S.B.;Li,Z.H.;Chen,C.Electrochim.Acta2010,55,7969.doi:10.1016/j.electacta.2010.03.025
(16) Kumar,A.;Singh,K.;Pandy,O.P.Journal of Refractory Metals and Hard Materials2011,29,555.doi:10.1016/j.ijrmhm.2011.01.009
(17) Reddy,K.M.;Rao,T.N.;Radha,K.;Joardar,J.Journal of Alloys and Compounds2010,494,404.doi:10.1016/j.jallcom.2010.01.059
(18) Zhou,X.S.;Qiu,Y.J.;Yin,J.;Gao,S.International Journal of Hydrogen Energy2011,36,7398.doi:10.1016/j.ijhydene.2011.03.081
(19) Rahsepar,M.;Pakshir,M.;Nikolaev,P.;Safavi,A.;Palanisamy,K.;Kim,H.Applied Catalysis B:Environmental2012,127,265.doi:10.1016/j.apcatb.2012.08.032
(20) Li,G.H.;Tian,W.;Tang,J.Y.;Ma,C.A.Acta Phys.-Chim.Sin.2007,23(9),1370.[李国华,田 伟,汤俊艳,马淳安.物理化学学报,2007,23(9),1370.]doi:10.3866/PKU.WHXB20070912
(21)Yao,G.X.;Shi,B.B.;Li,G.H.;Zheng,Y.F.Acta Phys.-Chim.Sin.2010,26(5),1317.[姚国新,施斌斌,李国华,郑遗凡.物理化学学报,2010,26(5),1317.]doi:10.3866/PKU.WHXB20100337
(22)Tauster,S.J.;Fung,S.C.;Garten,R.L.J.Am.Chem.Soc.1978,100,170.doi:10.1021/ja00469a029
(23)Hu,X.C.;Chen,D.;Shi,B.B.;Li,G.H.Acta Phys.-Chim.Sin.2011,27(12),2863.[胡仙超,陈 丹,施斌斌,李国华.物理化学学报,2011,27(12),2863.]doi:10.3866/PKU.WHXB20112863
(24)Li,G.H.;Chen,D.;Zheng,X.;Xie,W.M.;Chen,Y.Acta Phys.-Chim.Sin.2012,28(9),2077.[李国华,陈 丹,郑 翔,谢伟淼,程 媛.物理化学学报,2012,28(9),2077.]doi:10.3866/PKU.WHXB201206042
(25)Okamoto,H.;Kawamura,G.;Ishikawa,A.;Kudo,T.J.Electrochem.Soc.1987,134,1653.doi:10.1149/1.2100730
(26)Zhong,C.J.;Mathew,M.M.Adv.Mater.2001,13,1507.doi:10.1002/1521-4095(200110)13:19<>1.0.CO;2-S
(27) Cachet-Vivier,C.;Vivier,V.;Cha,C.S.;Nedlec,J.Y.;Yu,L.T.Electrochim.Acta2001,47,181.doi:10.1016/S0013-4686(01)00549-7
(28) Frelink,T.;Visscher,W.;van Veen,J.A.R.J.Electroanal.Chem.1995,382,65.doi:10.1016/0022-0728(94)03648-M
