磷酸化蛋白质组学的新进展及其在肝脏生理和病理机制中的应用
2014-06-15衣泰龙田苗苗杨晓明徐平
衣泰龙,田苗苗,杨晓明,徐平
综 述
磷酸化蛋白质组学的新进展及其在肝脏生理和病理机制中的应用
衣泰龙1,2,田苗苗2,杨晓明1,2,徐平1,2
1 安徽医科大学,安徽 合肥 230032 2 军事医学科学院放射与辐射医学研究所 北京蛋白质组研究中心蛋白质组学国家重点实验室,北京 102206
蛋白质磷酸化是最常见的蛋白质翻译后修饰形式。由于蛋白质的磷酸化形式可以被磷酸酶和磷酸激酶进行可逆的调控,所以在众多的生命活动过程中蛋白质的磷酸化修饰起着重要的调控作用,因此对生物体内蛋白质磷酸化修饰的系统研究对于揭示生命科学的奥秘显得十分重要。近年来,随着质谱技术和生物信息学软件以及磷酸化肽段富集方法的发展,利用质谱对生物体内蛋白质磷酸化修饰研究的技术逐渐成熟。肝脏作为人体最重要的代谢和免疫器官,深入研究肝脏细胞内蛋白质磷酸化修饰形式对于理解其功能具有重要指导意义。目前,迅速发展的磷酸化蛋白质组学技术已经被广泛应用到肝脏功能的生物学研究中。这些研究加深了人们对肝脏的生理及病理状态的分子生物学机制的了解。本文综述了当前磷酸化蛋白质组学的研究进展和磷酸化蛋白质组学在肝脏中的研究。
蛋白质磷酸化,肝脏磷酸化蛋白质组,磷酸化肽富集,定量蛋白质组,生物信息学
磷酸化修饰是蛋白质最主要的翻译后修饰形式之一。蛋白质的磷酸化修饰参与了细胞周期调控、细胞凋亡、发育、分化和疾病的发生等众多过程,对生命活动起着非常重要的调控作用。例如:线虫种系的发育[1]、神经突触囊泡的运输[2]、组织和器官特性的维持[3-4]、癌症的发生与发展[5-6]等几乎所有的生命活动过程和蛋白质的磷酸化修饰有着重要的关联。因此蛋白质磷酸化修饰的研究对于揭示生命的奥秘,疾病的诊断和治疗等有着非常重要的意义。
磷酸化蛋白质组学作为蛋白质组学的一部分内容,其研究技术归功于蛋白质组学技术的发展,包括质谱仪器的发展、样品的分离、定量技术的发展以及鉴定软件的开发。由于磷酸化蛋白质和磷酸化肽段的特殊性,对磷酸化蛋白质的研究需要一些特殊的手段。首先从化学计量的角度来讲,在生物体内磷酸化蛋白的丰度低于非磷酸化蛋白,因而在质谱检测时磷酸化肽段的信号会受到非磷酸化肽段信号的掩盖,因此在通常情况下很难直接用质谱检测到磷酸化肽段。为此,人们发展了一系列的磷酸化蛋白或磷酸化肽段特殊的染色方法、标记方法和富集方法,用于高灵敏度和特异性的磷酸化蛋白质的检测。此外,由于修饰的磷酸基团的存在,磷脂键的不稳定性使得磷酸化肽段在常规的碰撞诱导解离 (Collision induced association, CID) 检测时易产生信号很强的中性丢失峰,难以有效鉴定。为应对这种局面,不同的碎裂方式得以尝试,并取得了良好的结果。再辅以一系列针对磷酸化肽段鉴定和位点确定的质谱软件,使得磷酸化蛋白质组研究的技术日趋成熟,已经成为蛋白质翻译后修饰研究的典范。
成熟的磷酸化蛋白质组技术促进了生命科学研究的深入开展。肝脏是生物体重要的功能器官,负责机体的代谢、供能、体温维持和机体免疫等功能。随着磷酸化蛋白质组学技术的发展和成熟,肝脏相关的磷酸化蛋白质组学的研究逐渐增多。本文就磷酸化肽的分离和富集策略、磷酸化肽鉴定软件开发、磷酸化肽碎裂模式探索以及肝脏磷酸化蛋白质组学研究的新进展进行综述。
1 磷酸化肽的富集分离方法
在早期的磷酸化蛋白质组学研究中,通常使用双向凝胶电泳技术 (2-DE) 根据磷酸化蛋白与非磷酸化蛋白等电点的差异进行样品分离,再使用32P标记和磷酸化蛋白的特异染色鉴定磷酸化蛋白[7]。但是这两种策略都由于双向凝胶电泳技术本身的限制,难以满足大规模、高通量、高效率的磷酸化蛋白质组学研究的需要。
近来,适用于大规模磷酸化蛋白质组学研究的多肽样品分离和磷酸化肽高效富集方法爆炸式产生,促进了磷酸化蛋白质组学研究的快速发展。
磷酸化修饰肽段上的磷酸基团的存在,使得磷酸化肽与非磷酸化肽所带的电荷不同,所以最常用的样品初级分离方法是离子交换分离。为充分分离、简化或富集特定的组分,强阳离子交换色谱 (Strongcation exchange chromatography,SCX)[8]、强阴离子交换色谱(Strong anion exchange chromatography,SAX)[9]、多维液相色谱 (Multidimensional liquid chromatography)[10]、亲水相互作用色谱(Hydrophilic interaction liquid chromatography,HILIC)[11]、静电排斥-亲水性相互作用色谱(Electrostatic repulsion-hydrophilic interaction chromatography,ERLIC)[12]等多样的分离策略得到了深入研究和广泛应用。但离子交换色谱往往柱效比较低,且分离后的样品需要进行再次的脱盐操作。而近几年来发展的耐高pH的反相液相色谱 (High pH RP) 因其拥有高柱效,分离后的组分不需进行再次脱盐等优势,并且和蛋白质组学上样品检测使用的LC-MS/MS中的反相液相色谱的正交性较好而逐渐被应用于磷酸化蛋白质组学的研究中。
在磷酸化肽段的富集方面,适于抗体富集法[13]、固相金属离子亲和色谱 (Immobilized metal ion affinity chromatography,IMAC)[14]、金属氧化物亲和色谱 (Metal oxide affinity chromatography,MOAC)[15]等的亲和介质和富集技术得到了有效发展。由于这些方法通常都兼具磷酸化肽段分离和富集的作用,并具有一定的互补性,形成了丰富并各具特色的组合。而抗体、IMAC和MOAC等方法由于对磷酸化肽富集的高特异性,使得在大多数情况下将其他几种方法用作磷酸化样品的初分离,并形成了以亲疏水性和离子交换等为分离方式的IMAC富集方法[16-17]、以离子交换为分离方式的MOAC的富集方法[8]以及抗体富集与IMAC[18]和MOAC[19]结合等众多方法来实现磷酸化蛋白质组学的高覆盖。下面分别对磷酸化肽富集和分离的方法进行分析。
1.1 磷酸化肽的富集
现在常用的磷酸化肽的富集方法主要是IMAC和MOAC[20]。但是这两种方法均存在着非特异性吸附和吸附能力不足的缺陷。此外,针对磷酸化修饰的酪氨酸残基的抗体,也是磷酸化蛋白质组学研究中常用的富集材料[18]。表1中系统比较了这3种方法的优缺点。
在IMAC研究中,由于酸性基团 (如羧基等) 的存在,高丰度非磷酸化肽造成的非特异性吸附阻碍了磷酸化肽段的富集。Ficarro通过酯化反应来封闭样品中磷酸基团以外的其他酸性基团,这在一定程度上提高了IMAC富集的特异性。但由于该方法副反应产物的存在又限制了此方法的使用[14]。
除了非特异性吸附之外,吸附能力的大小也是一个影响富集效果的重要因素。系统比较不同的吸附离子后发现在Fe3+、Ga3+、Al3+、Zn2+、Cu2+、ZrO2+等诸多离子中,Fe3+的特异性最高,可达到93%。在此基础上还建立了以Fe3+为螯合介质的富集磷酸化肽段的优化流程[16-21]。我国科学家在磷酸化肽段的富集方面也做了大量的创新性工作。如中国科学院大连化学物理研究所的邹汉法团队对Fe3+、Ti4+和Zr4+富集磷酸化肽段的能力进行了系统研究,发现Ti4+和Zr4+对磷酸化肽的吸附性和特异性都要比Fe3+的好[22-27],这为磷酸化肽段富集技术的进一步发展奠定了基础。
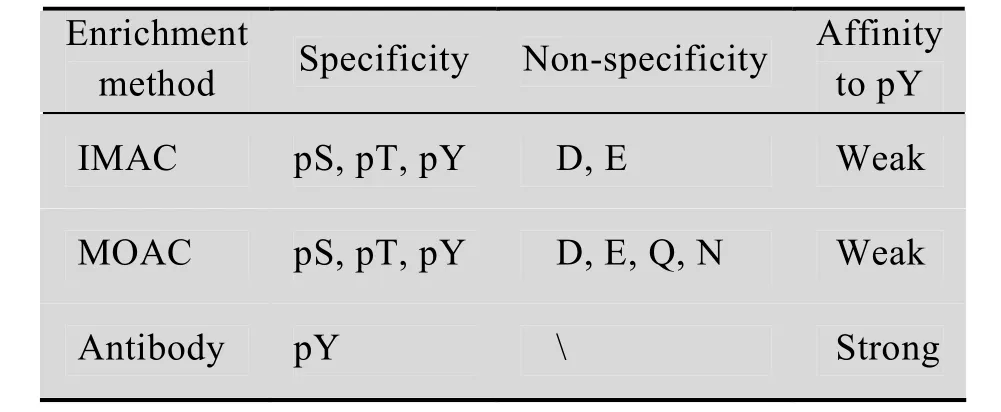
表1 不同类型的磷酸化肽富集方法的比较Table 1 Comparison of different types of phosphopeptides enrichment methods
除了金属离子的选择外,固相金属亲和介质的选择也是影响IMAC方法富集效率的重要因素。亚氨基二乙酸 (IDA)[14]、次氮基三乙酸(NTA)[17,28]是常用的金属螯合亲和介质。但是对于不同的吸附离子,最佳的金属螯合亲和介质是不同的。例如:在以Fe3+为吸附离子时磷酸化富集中利用NTA (富集效率:93%) 作介质的效果要优于IDA (富集效率:7.1%),而以ZrO2+为吸附离子时,以IDA为金属螯合亲和介质时的富集效率 (37%) 要高于以NTA为金属螯合亲和介质时的富集效率 (7.1%)[21]。最近邹汉法团队发现以共价键连接到载体上的磷酸基团作为配基的IMAC (Ti4+,Zr4+作为吸附离子) 在磷酸化肽富集的特异性和吸附能力方面都要比传统的Fe3+-IMAC的效果好[25-26]。
富集手法对磷酸化肽段的富集效果也有影响。Yue等发现通过多次富集可以增加大约18%的磷酸化肽段的鉴定数量。在富集的磷酸化肽段中,多磷酸化的肽段会优先被富集,但是丝氨酸、苏氨酸和酪氨酸修饰的磷酸化位点比例在3次富集中并没有明显差异[29]。
在MOAC富集磷酸化肽段的研究中,TiO2是最常用的金属氧化物。此外,硅−氧化镧等新的氧化物材料也得到了尝试[30]。与IMAC相似,酸性氨基酸的吸附也是影响磷酸化肽段富集特异性的一个重要因素。而在富集样品前使用二羟基苯甲酸 (DHB)[31-32]、天冬氨酸[33]和谷氨酸[20,34]等,可有效降低介质对富含酸性氨基酸肽段的非特异性吸附。除了对酸性氨基酸的非特异性吸附之外,Kanshin等还发现TiO2会对含谷氨酰胺和天冬酰胺肽的肽段也产生特异吸附,通过加入游离的天冬酰胺和谷氨酰胺可以降低80%非磷酸化肽的丰度[35]。
由于纳米材料具有较大的相对表面积,最近也被应用到了IMAC和MOAC中[20]。另外,特殊的纳米颗粒也为磷酸化肽的吸附提供了特殊的环境,使得磷酸化肽的结合速度也得到了提高。这些特点使得纳米材料在磷酸化蛋白质组学领域具有良好的应用前景[36]。然而,这些纳米材料的应用并没有改变原来的吸附原理,仍然是通过金属氧化物[37]或金属离子[38-42]对磷酸化肽进行富集。具有相似原理的高吸附能力的包被在石墨烯上的聚多巴胺 (Polydopamine)在磷酸化肽的富集方面也得到了应用[43]。
抗体在磷酸化蛋白质组研究中也得到了很好地应用。目前虽然市场上有针对丝氨酸、苏氨酸、酪氨酸等针对磷酸化修饰肽段的抗体,但前两者的特异性差,在一定程度上限制了抗体富集技术在磷酸化蛋白质组学上的应用。而针对酪氨酸磷酸化修饰肽段抗体的特异性好,可用于酪氨酸磷酸化蛋白质组学研究[44-46]。
因为不同方法在富集磷酸化肽时有很高的互补性[47],所以出现了一系列通过不同方法组合进行磷酸化肽富集的研究。例如,Tsai等通过Ga3+和Fe3+先后富集,发现各有41%和51%特异性富集的磷酸化肽[48]。
1.2 磷酸化肽的分离策略
为了降低磷酸化肽的复杂度,磷酸化肽的预分离显得很重要。理论上,可用于蛋白质组学的降低肽段复杂度的方法也可以用于磷酸化肽的分离。表2对不同分离原理的方法的优缺点进行了系统比较。
由于磷酸化肽所带的电荷数比未修饰的肽段少一个电荷,所以通过离子交换的方法进行的分离也起到了对磷酸化肽富集的作用[16]。同时,磷酸基团作为亲水基团还改变了磷酸化肽的亲疏水性。所以根据亲疏水性进行的分离方法同样也具有磷酸化肽富集的效果[11-12,17]。
SDS-PAGE作为常用的蛋白质分离方法,可根据分子量大小对蛋白质进行分离。然而,磷酸化蛋白质更多地出现在高分子量区域[49]。而且,SDS-PAGE分离蛋白质的载样量相对较小,所以现在很少用这种方法。
不论是根据亲疏水性还是通过离子交换对肽段进行分离,虽然都有效地降低了磷酸化肽的复杂度,但磷酸化肽本身并没有得到很好的分离。而且有研究比较发现,在相同机时的情况下,通过二维分离可以比一维分离多鉴定40%的磷酸化肽段[50];Mohammed实验室发现,在样品量足够 (不同的样品,所需的起始材料的量不同) 的情况下,通过三维液相分离鉴定的磷酸化位点比通过二维液相分离鉴定的磷酸化位点多了29% (多鉴定4 092个磷酸化位点)[51]。
最近Carr实验室通过高pH反相液相色谱分离,并采取每隔24个组分进行合并的方式,实现了磷酸化肽的平均分配并且单次实验最多鉴定到了超过22 000个磷酸化位点[17]。
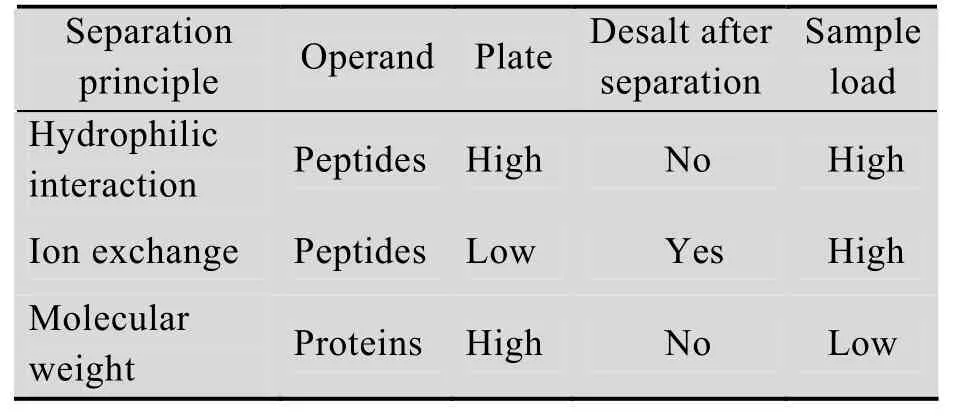
表2 不同原理的蛋白/肽段分离方法的比较Table 2 Comparison of protein/peptide separation methods based on different separation principle
2 磷酸化肽段的鉴定
2.1 磷酸化肽段的碎裂和鉴定方式
目前常用的磷酸化肽段碎裂方式主要有碰撞诱导解离 (Collision induced dissociation,CID)、电子捕获解离 (Electron capture dissociation, ECD)、电子转运解离 (Electron transfer dissociation, ETD) 和高能碰撞解离(Higher energy collision dissociation, HCD) 等。表3对3种碎裂方式在磷酸化蛋白质组学应用中的优缺点进行了系统比较。
CID因是绝大多数质谱仪的标准配置,具有扫描速度快、灵敏度高等诸多优点,因此和蛋白质鉴定一样,在磷酸化蛋白质组中得到了广泛的应用。但是,在CID碎裂时易发生磷酸基团的中性丢失,导致碎裂不完全,因而二级谱图的质量差。尽管中性丢失基团可以作为磷酸化肽段的标志,还可通过激发中性丢失离子的第三级碎裂 (MS3或pseudo-MS3碎裂) 增加鉴定可靠性[52-53]。但是,这种方式并不能提高磷酸化肽段的鉴定量,而且对于确定磷酸化位点的贡献也比较小[54]。此外,通过CID进行碎裂的过程中磷酸化基团还易发生分子内转移,增加了鉴定的难度[55-56]。
为了解决CID碎裂过程中的中性丢失问题,Zubarev和Kelleher发明的ECD和美国维杰尼亚大学Hunt实验室Coon等发明的ETD先后被用于磷酸化肽段的鉴定。这两种碎裂模式均可使磷酸化肽中的磷脂键在碎裂过程中保持较好的完整性,二级谱图质量明显提高,极大地增加了磷酸化肽位点的可靠性[20]。Pandey实验室比较了ETD和CID在磷酸化肽鉴定的效果,发现用ETD比用CID多了40%肽段碎裂离子,由此多鉴定了60%的磷酸化肽[57]。
HCD由于高能碰撞,碎裂更加充分,可以产生更多的碎裂离子。这使得二级谱图的质量更高,可得到可信度更高的鉴定结果[58],因而在磷酸化肽段鉴定中逐渐得到了更多的应用。尽管如此,Gygi实验室通过‘back-to-back’的方法严格地比较了CID和HCD在鉴定磷酸化肽段中的效果,发现通过HCD碎裂得到的谱图的XCorr、Δ Cn和Ascore值比用CID碎裂得到的高,但是由于HCD的扫描速度比CID慢,因此在相同的条件下,CID可得到更多的磷酸化肽段的鉴定[18]。与ETD相比,Kuster实验室通过大规模合成磷酸化肽发现HCD的鉴定能力超过了ETD[59]。随着仪器的发展,扫描速度的进一步加快,可以预见HCD会在磷酸化肽的鉴定中具有更广的应用前景。
除了上述几种碎裂方式外,Mohammed实验室新近发展的电子转移高能碰撞裂解(Electron-transfer/higher-energy collision dissociation, EThcD) 可实现肽段的ETD和HCD的级联碎裂,使得磷酸化肽可以产生c-、z-、b-和y-离子。这些更为丰富的肽段碎裂离子在磷酸化位点的确定中具有更好的指示作用,显现了优于HCD和ETD的性能[60]。
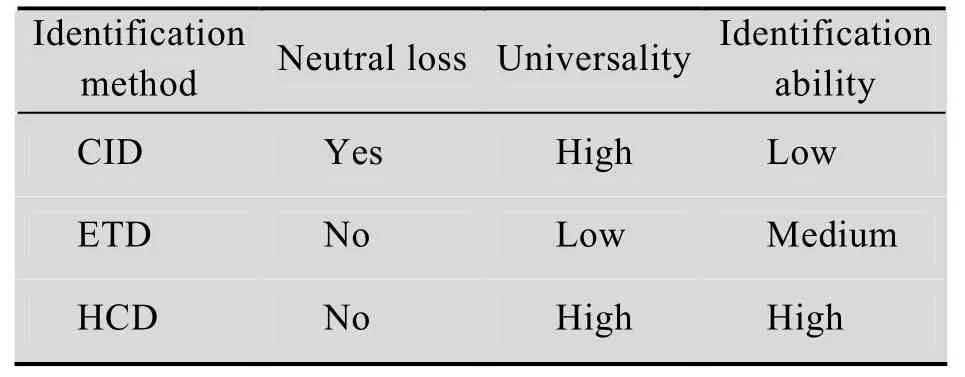
表3 常用磷酸化肽段碎裂鉴定方法的比较Table 3 Comparison of commonly used phosphopeptides dissociation methods
2.2 磷酸化肽的鉴定和位点确定软件的开发
磷酸化肽段的鉴定中常用的软件有Sequest、Mascot和Mann实验室新近开发的Andromeda等[61]。Thermo公司组合了Sequest和Mascot等搜索引擎,形成了ProteomeDiscoverer,并在蛋白质和磷酸化蛋白质的鉴定中也得到了应用。在国内,中国科学院计算技术研究所的贺思敏团队开发了pFind系列软件,并在磷酸化蛋白质组研究中得到了测试,显示了良好的性能[62]。值得注意的是,除Andromeda和pFind为免费搜索引擎外,其他的都需要付费才能使用。
在磷酸化肽的鉴定中,位点的确定是一个难题。目前主要的位点校正软件主要有Gygi实验室用在Sequest搜索结果上的Ascore[63]、Mann实验室用于Mascot和Andromeda引擎检索结果后的PTM score[8,59,61]、Thermo公司Proteome Discoverer中的phosphoRS和用于多级碎裂的PhosphoScore[64]以及Mascot Delta Score[59,65]等。D-score是一个最近开发的用于磷酸化位点校正的软件,可以用于Mascot、OMSSA和X!Tandem等的鉴定结果[66]。目前对于不同的软件还没有比较研究的结果发表。
最近利用大规模合成的磷酸化肽段评估质谱鉴定的结果,并比较了鉴定软件Andromeda、Mascot以及磷酸基团定位软件MD score、PTM score和PhosphoRS的性能。结果发现,在以decoy的方法进行质控的时候,肽段鉴定结果的FDR和鉴定的磷酸化位点的FDR都有低估现象,并发现这些软件在鉴定等方面具有一定的互补性[59]。
3 定量磷酸化蛋白质组学研究
磷酸化蛋白质组的定量与普通的蛋白质组的定量类似,都是直接通过肽段水平的定量实现的。所以,应用于蛋白质组学的定量技术也都可以应用于磷酸化的定量。但由于同一肽段不同位点磷酸化变化并不一定一致[8],因此,磷酸化蛋白质组的定量是蛋白质具体肽段的定量值,而非来自同一个蛋白的不同肽段的定量值的整合。在实验过程中可能引入的误差,如蛋白质消化过程中的漏切、相同肽段可能存在的多个修饰位点的情况等都使得磷酸化位点的直接定量难度较大。因此研究具体的磷酸化位点的变化的时候需要人工仔细检查。
已有的用于蛋白质组学的定量技术,如SILAC、iTRAQ和TMT等均可以用于磷酸化定量。其中,iTRAQ和TMT等化学标记定量结果通常会因诸多技术原因缩小了自然状态的差异,尽管这种差异可通过MS3和气相净化(Gas-phase purification) 技术得以部分克服[67-68],但是磷酸化研究中所用的起始材料量往往比较大,不仅化学标记本身的成本是一个限制因素,而且至今还鲜有通过MS3和气相净化进行定量的报道。最近,邹汉法实验室通过还原二甲基化作用引入稳定同位素的方法被用于磷酸化蛋白质组学的定量研究中[69]。但是引入的同位素保留时间的不一致可能会影响定量的准确性[70]。
在磷酸化研究中,实验组与对照组之间磷酸化修饰的相对变化是生物学研究中关注的主要内容。除此之外,为了研究一个磷酸化位点在生物体内的修饰与未修饰的比例,Gygi实验室通过还原二甲基化作用发展了一种研究磷酸化与未磷酸化比例的定量方法[71]。
4 日趋成熟的磷酸化蛋白质组学技术
随着磷酸化肽段的富集、分离和鉴定技术的日趋成熟和质谱仪器的快速发展,产生的磷酸化蛋白质组学的数据规模逐渐增大,磷酸化蛋白质组学的研究技术已趋于成熟。例如,Gygi实验室通过研究不同的小鼠组织和器官,共鉴定到了将近36 000个磷酸化位点[4]。Olsen通过研究小鼠的不同器官和组织,共鉴定到了31 480个位点[3]。这是当前最大的磷酸化蛋白质组学数据集。单次实验鉴定的数量也在不断增多,并已经超过20 000个位点[17]。与此同时,进行磷酸化研究所需要的总样品量也在减少。最近Zhou等用500 μg蛋白的消化产物鉴定到了13 681个磷酸化位点,3 mg蛋白消化产物鉴定到了18 055个磷酸化位点[51]。此外,有研究还发现通过EDTA处理液相色谱系统的管路,可以降低金属管路对磷酸化肽,尤其是多个磷酸化位点修饰的磷酸化肽的吸附,从而有效地提高了磷酸化肽段的覆盖度[72]。
随着对磷酸化研究的不断深入,预测激酶底物的软件iPGS[73]、构建磷酸化信号通路的软件Answer Set Programming[74]和Sorad[75]等得以成功开发。磷酸化蛋白质组学实验技术和生物信息学工具的发展将为复杂的生命活动规律的揭示提供有效手段[3,8,76]。
5 肝脏磷酸化蛋白质组学研究
肝脏涉及到食物的消化、能量的供应、体温的维持、免疫调节、解毒以及许多重要代谢中间体的合成等诸多功能,是高等生物机体内新陈代谢最活跃的器官。肝脏生物学功能的失调会导致机体严重的疾病,甚至死亡,因此肝脏的生理和病理研究长期以来一直是健康医学研究的热点。蛋白质磷酸化参与了生物体内几乎所有的生命活动过程。这在肝脏生理和病理蛋白质组学研究中也得到了充分的体现。
在肝脏疾病模型的磷酸化蛋白质组学研究方面,糖尿病是受到较多关注的方向。糖尿病与胰岛素相关的信号通路密切相关,已有研究表明在胰岛素刺激过程中,包括Akt和PI3K/mTOR在内的超过5 000个磷酸化位点会发生修饰水平的变化[77]。因此磷酸化蛋白质组学的研究对于揭示糖尿病的致病机制具有重要的指导意义。曾嵘实验室对早期发病阶段的二型糖尿病肝脏线粒体进行了包括磷酸化蛋白质组在内的多个层面的蛋白质组学的研究[78]。Pshezhetsky实验室发现胰岛素会引发囊泡运输、代谢、细胞运动和基因表达等众多过程的蛋白质的磷酸化的变化[79]。Pagliarini实验室通过对肥胖和二型糖尿病小鼠模型肝脏线粒体进行研究,发现磷酸化在酮体生成过程中起着重要作用,并发现催化酮体生成的限速酶Hmgcs2 (S456)的磷酸化修饰可以提高其催化酮体生成的活性[80]。
肝脏也是重要的解毒器官。Dai实验室通过对磷酸化蛋白质组学研究发现全氟十二酸会抑制胰岛素信号通路,并推测脂质体的升高抑制了糖原合酶的激酶的活性[81]。这一研究将为肝脏解毒的机制研究提供磷酸化修饰方面的基础。但是因为这一研究只鉴定了1 443个磷酸化位点,所以深入的大规模的研究还有待开展。
肝癌是威胁人类健康的重大隐患。目前的研究发现,在肝癌转移过程中RAF/mitogenactiv atedprotein kinase (MAPK) /extracellular signal-regulated kinase (ERK) kinase (MEK)/ ERK (RAF/MEK/ERK) 等与磷酸化密切相关的信号通路通常处于活化状态,这说明蛋白质的磷酸化修饰与肝癌的侵袭转移机制密切相关[82-83]。因此对于肝癌的磷酸化蛋白质组学的研究显得十分重要。目前以肝癌为模型的磷酸化蛋白质组学研究还比较少。邹汉法实验室[68]定量比较研究了正常肝脏组织和肝癌组织的磷酸化差异,发现肝癌中ERK信号通路中众多蛋白质的磷酸化水平发生了变化,例如B-Raf的S729位点磷酸化水平上调了两倍。Lee等[84]通过定量比较正常和肝癌细胞磷酸化蛋白质组的差异,筛选并确定了潜在的肝癌生物标志物。Mann实验室通过定量比较磷酸酶抑制剂对小鼠肝脏细胞系Hepa1-6磷酸化影响,发现只有很少的磷酸化肽的修饰受到了明显的影响。这也暗示着更有效的磷酸酶抑制剂需要用于磷酸化蛋白质组学样品的收集和制备中[19]。
肝脏生理条件下蛋白质磷酸化状态及其动态变化规律是理解肝脏功能及其调控的基础,但至今这方面的研究也还比较少。Gygi实验室对小鼠肝脏磷酸化肽进行了基序和位点分析,发现了直接与脯氨酸相邻的基序、碱性基序、酸性基序和含有碱性氨基酸和酸性氨基酸的双极性基序等,并且发现磷酸化位点更容易出现在蛋白质的羧基末端 (C端)[85]。这为预测和寻找激酶底物提供了依据,也为分析不同的磷酸化肽富集方法的偏好性提供了方法和工具。此外,Gygi实验室还分析了小鼠包括肝脏在内的9个不同器官的磷酸化修饰,发现了3%的肝脏特异性的磷酸化位点,并发现了Mek2等在肝脏和肺等特异的磷酸化修饰[4]。在Gygi实验室之后,Olsen实验室也定量比较了包括肝脏在内的不同器官和组织的磷酸化蛋白质组的差异,发现糖原分解酶的酪氨酸位点在骨骼肌中被磷酸化,但是在肝脏中却并没有被磷酸化[3]。这为人们理解不同组织的生物学功能以及肝脏的磷酸化状态提供了依据。Gygi实验室还用在低pH条件下还原二甲基化作用的定量标记方法研究了禁食和重新喂食小鼠肝脏磷酸化蛋白质组学的变化,并观察到了RSK2在重新喂食2 h后被磷酸化激活等新的发现[70]。中国科学院大连化学物理研究所的邹汉法和华中科技大学的薛宇合作开发了iGPS软件,并对人肝脏激酶底物进行了预测,产生了当时最大的人类肝脏磷酸化蛋白质组学质谱数据集[73]。这一数字在2013年又被邹汉法实验室的22 446个磷酸化位点更新[85]。
6 展望
运动是生命形式的本质特征。生物体通过蛋白质不同位点、不同翻译后修饰形式或者同一类型的不同结构的翻译后修饰实现信号的接受、传递、放大和效应。因此蛋白质磷酸化的动态变化研究将是在成熟的磷酸化蛋白质组技术基础上的必然选择。随着翻译后修饰蛋白质组学研究方法的不断成熟,不同的翻译后修饰的串话研究正在成为研究热点。目前在蛋白质组学领域,与磷酸化串话的翻译后修饰的研究主要是泛素化、糖基化和乙酰化。
在磷酸化与乙酰化的串话研究中,Zhao实验室和Volchenboum实验室通过生物信息的方法预测乙酰化很有可能与包括磷酸化在内的众多修饰有串话[87];Bork实验室和Gavin实验室通过敲除两个激酶和一个磷酸酶发现了磷酸化和乙酰化修饰都发生了变化,并推测了潜在的串话[88]。
除了与乙酰化的串话之外,磷酸化与泛素化的串话也是研究的重点。Beltrao实验室和Krogan实验室整合了11种真核生物的磷酸化位点、泛素化位点和乙酰化位点,并发明了一种可以排列不同修饰的功能关联优先级的方法[89]。
此外,糖基化和磷酸化之间的串话是最近研究的比较多的另一种串话。Sze实验室通过同时研究鼠肾组织的蛋白质组、磷酸化蛋白质组和糖蛋白质组,发现多能蛋白聚糖核心蛋白(Versican core protein) 和纤连蛋白 (Fibronectin)等可以同时被磷酸化和糖基化修饰,暗示了两种修饰之间的串话[90]。Burlingame实验室通过对鼠突触进行糖基化和磷酸化蛋白质组学研究,发现激酶通常会被糖基化修饰,暗示着糖基化和磷酸化的串话是通过糖基化的修饰来调节激酶的活性实现的[91]。Yang实验室和Smitha实验室通过研究小鼠大脑的糖蛋白质组发现很多发生糖基化的位点靠近已知的磷酸化位点,这也支持蛋白质糖基化和磷酸化修饰之间的串话[92]。
此外,Bork实验室构建了PTMcode数据库,这个数据库包括了已知的和预测的磷酸化修饰等在内的不同翻译后修饰之间的功能的相互关系信息[93]。Nature Methods今年同时发表了两篇研究磷酸化和泛素化等修饰之间串话研究方法的论文[17,28],为众多翻译后修饰之间的串话的研究提供了思路。
目前对于肝脏尤其是肝癌的磷酸化蛋白质组学的生物学研究还比较少。虽有研究表明肝癌发生发展过程中RAF/mitogenactivated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase (MEK)/ERK (RAF/MEK/ERK)磷酸化通路会发生失调,但是其磷酸化位点的研究尚不充分,所以对磷酸化位点的研究将会成为肝癌未来研究的主要方向。随着磷酸化蛋白质组研究的深入,产生的大量数据需要生物信息学来解决,因此生物信息学在蛋白质组中的应用也将会成为磷酸化蛋白质组研究中不可缺少的一个重要环节。同时激酶抑制剂在肝癌发生或发展过程中对其磷酸化蛋白质组的影响将会成为筛选抗癌药物的有利手段。亚细胞器或者靶向的相互作用磷酸化蛋白质组学的研究将会成为将来深入探讨磷酸化作用机制的重要手段。随着技术的成熟以及生物研究的需要,对肝脏的磷酸化蛋白质组学的研究将会为揭示肝脏生物学的奥秘提供支持。
REFERENCES
[1] Arur S, Ohmachi M, Nayak S, et al. Multiple Erk substrates execute single biological processes in caenorhabditis elegans germ-line development. Proc Natl Acad Sci USA, 2009, 106(12): 4776–4781.
[2] Armbruster M, Messa M, Ferguson SM, et al. Dynamin phosphorylation controls optimization of endocytosis for brief action potential bursts. Elife, 2013, 2: e00845.
[3] Lundby A, Secher A, Lage K, et al. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun, 2012, 3: 876.
[4] Huttlin EL, Jedrychowski MP, Elias JE, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell, 2010, 143(7): 1174–1189.
[5] Ma WL, Hsu CL, Yeh CC, et al. Hepatic androgen receptor suppresses hepatocellular carcinoma metastasis through modulation of cell migration and anoikis. Hepatology, 2012, 56(1): 176–185.
[6] Tomasi ML, Tomasi I, Ramani K, et al. S-adenosyl methionine regulates ubiquitin-conjugating enzyme 9 protein expression and sumoylation in murine liver and human cancers. Hepatology, 2012, 56(3): 982–993.
[7] Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol, 2003, 21(3): 255–261.
[8] Olsen JV, Blagoev B, Gnad F, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell, 2006, 127(3): 635–648.
[9] Dong M, Ye M, Cheng K, et al. Depletion of acidic phosphopeptides by SAX to improve the coverage for the detection of basophilic kinase substrates. J Proteome Res, 2012, 11(9): 4673–4681.
[10] Albuquerque CP, Smolka MB, Payne SH, et al. Amultidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics, 2008, 7(7): 1389–1396.
[11] McNulty DE, Annan RS. Hydrophilic interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol Cell Proteomics, 2008, 7(5): 971–980.
[12] Alpert AJ. Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal Chem, 2008, 80(1): 62–76.
[13] Macek B, Mann M, Olsen JV. Global and site-specific quantitative phosphoproteomics: principles and applications. Annu Rev Pharmacol Toxicol, 2009, 49: 199–221.
[14] Ficarro SB, McCleland ML, Stukenberg PT, et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol, 2002, 20(3): 301–305.
[15] De Corte V, Demol H, Goethals M, et al. Identification of Tyr438 as the major in vitro c-Src phosphorylation site in human gelsolin: a mass spectrometric approach. Protein Sci, 1999, 8(1): 234–241.
[16] Villen J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc, 2008, 3(10): 1630–1638.
[17] Mertins P, Qiao JW, Patel J, et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods, 2013, 10(7): 634–637.
[18] Jedrychowski MP, Huttlin EL, Haas W, et al. Evaluation of HCD- and CID-type fragmentation within their respective detection platforms for murine phosphoproteomics. Mol Cell Proteomics, 2011, 10(12): M111.009910.
[19] Pan C, Gnad F, Olsen JV, et al. Quantitative phosphoproteome analysis of a mouse liver cell line reveals specificity of phosphatase inhibitors. Proteomics, 2008, 8(21): 4534–4546.
[20] Wang F, Song C, Cheng K, et al. Perspectives of comprehensive phosphoproteome analysis using shotgun strategy. Anal Chem, 2011, 83(21): 8078–8085.
[21] Ficarro SB, Adelmant G, Tomar MN, et al. Magnetic bead processor for rapid evaluation and optimization of parameters for phosphopeptide enrichment. Anal Chem, 2009, 81(11): 4566–4575.
[22] Zou HF, Ye M, Dong J, et al. Specific phosphopeptide enrichment with immobilized titanium ion affinity chromatography adsorbent for phosphoproteome analysis. J Proteome Res, 2008, 7(9): 3957–3967.
[23] Yu Z, Han G, Sun S, et al. Preparation of monodisperse immobilized Ti(4+) affinity chromatography microspheres for specific enrichment of phosphopeptides. Anal Chim Acta, 2009, 636(1): 34–41.
[24] Zou HF, Low TY, Hennrich ML, et al. Enhancing the identification of phosphopeptides from putative basophilic kinase substrates using Ti (IV) based IMAC enrichment. Mol Cell Proteomics, 2011, 10(10): M110. 006452.
[25] Zou HF, Ye M, Dong J, et al. Robust phosphoproteome enrichment using monodisperse microsphere-based immobilized titanium (IV) ion affinity chromatography. Nat Protoc, 2013, 8(3): 461–480.
[26] Feng S, Ye M, Zou HF, et al. Immobilized zirconium ion affinity chromatography for specific enrichment of phosphopeptides in phosphoproteome analysis. Mol Cell Proteomics, 2007, 6(9): 1656–1665.
[27] Qi D, Mao Y, Lu J, et al. Phosphate-functionalized magnetic microspheres for immobilization of Zr(4+) ions for selective enrichment of the phosphopeptides. J Chromatogr A, 2010, 1217(16): 2606–2617.
[28] Swaney DL, Beltrao P, Starita L, et al. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat Methods, 2013, 10(7): 676–682.
[29] Yue XS, Hummon AB. Combination of multistep IMAC enrichment with high-pH reverse phase separation for in-depth phosphoproteomic profiling. J Proteome Res, 2013, 12(9): 4176–4186.
[30] Jabeen F, Hussain D, Fatima B, et al. Silica-lanthanum oxide: pioneer composite of rare-Earth metal oxide in selective phosphopeptides enrichment. Anal Chem, 2012, 84(23): 10180–10185.
[31] Thingholm TE, Jorgensen TJ, Jensen ON, et al.Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat Protoc, 2006, 1(4): 1929–1935.
[32] Mazanek M, Mituloviae G, Herzog F, et al. Titanium dioxide as a chemo-affinity solid phase in offline phosphopeptide chromatography prior to HPLC-MS/MS analysis. Nat Protoc, 2007, 2(5): 1059–1069.
[33] Chi M, Bi W, Lu Z, et al. Application of aspartic acid as a non-specific binding inhibitor in the enrichment of phosphopeptides with titanium dioxide. Se Pu, 2010, 28(2): 152–157 (in Chinese).迟明, 毕炜, 卢庄, 等. 天冬氨酸作为非特异性吸附抑制剂在二氧化钛选择性富集磷酸肽中的应用. 色谱, 2010, 28(2): 152–157.
[34] Wu J, Shakey Q, Liu W, et al. Global profiling of phosphopeptides by titania affinity enrichment. J Proteome Res, 2007, 6(12): 4684–4689.
[35] Kanshin E, Michnick SW, Thibault P. Displacement of N/Q-rich peptides on TiO2beads enhances the depth and coverage of yeast phosphoproteome analyses. J Proteome Res, 2013, 12(6): 2905–2913.
[36] Han L, Shan Z, Chen D, et al. Mesoporous Fe2O3microspheres: rapid and effective enrichment of phosphopeptides for MALDI-TOF MS analysis. J Colloid Interface Sci, 2008, 318(2): 315–321.
[37] Zhou H, Tian R, Ye M, et al. Highly specific enrichment of phosphopeptides by zirconium dioxide nanoparticles for phosphoproteome analysis. Electrophoresis, 2007, 28(13): 2201–2215.
[38] Pan C, Ye M, Liu Y, et al. Enrichment of phosphopeptides by Fe3+-immobilized mesoporous nanoparticles of MCM-41 for MALDI and nano-LC-MS/MS analysis. J Proteome Res, 2006, 5(11): 3114–3124.
[39] Wei J, Zhang Y, Wang J, et al. Highly efficient enrichment of phosphopeptides by magnetic nanoparticles coated with zirconium phosphonate for phosphoproteome analysis. Rapid Commun Mass Spectrom, 2008, 22(7): 1069–1080.
[40] Zhao L, Wu R, Han G, et al. The highly selective capture of phosphopeptides by zirconium phosphonate-modified magnetic nanoparticles for phosphoproteome analysis. J Am Soc Mass Spectrom, 2008, 19(8): 1176–1186.
[41] Zhao L, Wu R, Zou HF. Selective capture of phosphopeptides by zirconium phosphonate-magnetic nanoparticles. Methods Mol Biol, 2011, 790: 215–222.
[42] Zhong H, Xiao X, Zheng S, et al. Mass spectrometric analysis of mono- and multi-phosphopeptides by selective binding with NiZiZnFe2O4magnetic nanoparticles. Nat Commun, 2013, 4: 1656.
[43] Yan Y, Zheng Z, Deng C, et al. Hydrophilic polydopamine-coated graphene for metal ion immobilization as a novel immobilized metal ion affinity chromatography platform for phosphoproteome analysis. Anal Chem, 2013, 85(18): 8483–8487.
[44] Wisniewski JR, Nagaraj N, Zougman A, et al. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res, 2010, 9(6): 3280–3289.
[45] Iliuk AB, Martin VA, Alicie BM, et al. In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol Cell Proteomics, 2010, 9(10): 2162–2172.
[46] Ruperez P, Gago-Martinez A, Burlingame AL, et al. Quantitative phosphoproteomic analysis reveals a role for serine and threonine kinases in the cytoskeletal reorganization in early T cell receptor activation in human primary T cells. Mol Cell Proteomics, 2012, 11(5): 171–186.
[47] Bodenmiller B, Mueller LN, Mueller M, et al. Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods, 2007, 4(3): 231–237.
[48] Tsai CF, Hsu CC, Hung JN, et al. Sequential phosphoproteomic enrichment through complementary metal-directed immobilized metal ion affinity chromatography. Anal Chem, 2013, 86(1): 685–693.
[49] Li X, Gerber SA, Rudner AD, et al. Large-scale phosphorylation analysis of alpha-factor-arrested Saccharomyces cerevisiae. J Proteome Res, 2007, 6(3): 1190–1197.
[50] Hennrich ML, Groenewold V, Kops GJ, et al. Improving depth in phosphoproteomics by using a strong cation exchange-weak anion exchange-reversed phase multidimensional separation approach. Anal Chem, 2011, 83(18):7137–7143.
[51] Zhou H, Di PS, Preisinger C, et al. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res, 2013, 12(1): 260–271.
[52] Beausoleil SA, Jedrychowski M, Schwartz D, et al. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA, 2004, 101(33): 12130–12135.
[53] Schroeder MJ, Shabanowitz J, Schwartz JC, et al. A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal Chem, 2004, 76(13): 3590–3598.
[54] Villen J, Beausoleil SA, Gygi SP. Evaluation of the utility of neutral-loss-dependent MS3 strategies in large-scale phosphorylation analysis. Proteomics, 2008, 8(21): 4444–4452.
[55] Palumbo AM, Reid GE. Evaluation of gas-phase rearrangement and competing fragmentation reactions on protein phosphorylation site assignment using collision induced dissociation-MS/MS and MS3. Anal Chem, 2008, 80(24): 9735–9747.
[56] Cui L, Reid GE. Examining factors that influence erroneous phosphorylation site localization via competing fragmentation and rearrangement reactions during ion trap CID-MS/MS and -MS(3.). Proteomics, 2013, 13(6): 964–973.
[57] Molina H, Horn DM, Tang N, et al. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci USA, 2007, 104(7): 2199–2204.
[58] Olsen JV, Macek B, Lange O, et al. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods, 2007, 4(9): 709–712.
[59] Marx H, Lemeer S, Schliep JE, et al. A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat Biotechnol, 2013, 31(6): 557–564.
[60] Frese CK, Zhou H, Taus T, et al. Unambiguous phosphosite localization using electron-transfer/ higher-energy collision dissociation (EThcD). J Proteome Res, 2013, 12(3): 1520–1525.
[61] Cox J, Neuhauser N, Michalski A, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res, 2011, 10(4): 1794–1805.
[62] Fu Y, Jia W, Lu Z, et al. Efficient discovery of abundant post-translational modifications and spectral pairs using peptide mass and retention time differences. BMC Bioinformatics, 2009, 10 (Suppl 1): S50.
[63] Beausoleil SA, Villen J, Gerber SA, et al. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol, 2006, 24(10): 1285–1292.
[64] Ruttenberg BE, Pisitkun T, Knepper MA, et al. PhosphoScore: an open-source phosphorylation site assignment tool for MSn data. J Proteome Res, 2008, 7(7): 3054–3059.
[65] Savitski MM, Lemeer S, Boesche M, et al. Confident phosphorylation site localization using the Mascot Delta Score. Mol Cell Proteomics, 2011, 10(2): M110 003830.
[66] Vaudel M, Breiter D, Beck F, et al. D-score: a search engine independent MD-score. Proteomics, 2013, 13(6): 1036–1041.
[67] Ting L, Rad R, Gygi SP, et al. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods, 2011, 8(11): 937–940.
[68] Wenger CD, Lee MV, Hebert AS, et al. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat Methods, 2011, 8(11): 933–935.
[69] Song C, Wang F, Ye M, et al. Improvement of the quantification accuracy and throughput for phosphoproteome analysis by a pseudo triplex stable isotope dimethyl labeling approach. Anal Chem, 2011, 83(20): 7755–7762.
[70] Wilson-Grady JT, Haas W, Gygi SP. Quantitative comparison of the fasted and re-fed mouse liver phosphoproteomes using lower pH reductive dimethylation. Methods, 2013, 61(3): 277–286.
[71] Wu R, Haas W, Dephoure N, et al. A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat Methods, 2011, 8(8): 677–683.
[72] Fleitz A, Nieves E, Madrid-Aliste C, et al. Enhanced detection of multiply phosphorylated peptides and identification of their sites of modification. Anal Chem, 2013, 85(18): 8566–8576.
[73] Song C, Ye M, Liu Z, et al. Systematic analysis ofprotein phosphorylation networks from phosphoproteomic data. Mol Cell Proteomics, 2012, 11(10): 1070–1083.
[74] Guziolowski C, Videla S, Eduati F, et al. Exhaustively characterizing feasible logic models of a signaling network using Answer Set Programming. Bioinformatics, 2013, 29(18): 2320–2326.
[75] Aijo T, Granberg K, Lahdesmaki H. Sorad: a systems biology approach to predict and modulate dynamic signaling pathway response from phosphoproteome time-course measurements. Bioinformatics, 2013, 29(10): 1283–1291.
[76] Zanivan S, Meves A, Behrendt K, et al. In vivo SILAC-based proteomics reveals phosphoproteome changes during mouse skin carcinogenesis. Cell Rep, 2013, 3(2): 552–566.
[77] Humphrey SJ, Yang G, Yang P, et al. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab, 2013, 17(6): 1009–1020.
[78] Deng WJ, Nie S, Dai J, et al. Proteome, phosphoproteome, and hydroxyproteome of liver mitochondria in diabetic rats at early pathogenic stages. Mol Cell Proteomics, 2010, 9(1): 100–116.
[79] Fedjaev M, Parmar A, Xu Y, et al. Global analysis of protein phosphorylation networks in insulin signaling by sequential enrichment of phosphoproteins and phosphopeptides. Mol Biosyst, 2012, 8(5): 1461–1471.
[80] Grimsrud PA, Carson JJ, Hebert AS, et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab, 2012, 16(5): 672–683.
[81] Zhang H, Hou J, Cui R, et al. Phosphoproteome analysis reveals an important role for glycogen synthase kinase-3 in perfluorododecanoic acid-induced rat liver toxicity. Toxicol Lett, 2013, 218(1): 61–69.
[82] Gauthier A, Ho M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: an update. Hepatol Res, 2013, 43(2): 147–154.
[83] Liu L, Cao Y, Chen C, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res, 2006, 66(24): 11851–11858.
[84] Lee HJ, Na K, Kwon MS, et al. Quantitative analysis of phosphopeptides in search of the disease biomarker from the hepatocellular carcinoma specimen. Proteomics, 2009, 9(12): 3395–3408.
[85] Villen J, Beausoleil SA, Gerber SA, et al. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci USA, 2007, 104(5): 1488–1493.
[86] Bian Y, Song C, Cheng K, et al. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J Proteomics, 2014, 96: 253–262.
[87] Lu Z, Cheng Z, Zhao Y, et al. Bioinformatic analysis and post-translational modification crosstalk prediction of lysine acetylation. PLoS ONE, 2011, 6(12): e28228.
[88] van Noort V, Seebacher J, Bader S, et al. Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol Syst Biol, 2012, 8: 571.
[89] Beltrao P, Albanese V, Kenner LR, et al. Systematic functional prioritization of protein posttranslational modifications. Cell, 2012, 150(2): 413–425.
[90] Hao P, Guo T, Sze SK. Simultaneous analysis of proteome, phospho- and glycoproteome of rat kidney tissue with electrostatic repulsion hydrophilic interaction chromatography. PLoS ONE, 2011, 6(2): e16884.
[91] Trinidad JC, Barkan DT, Gulledge BF, et al. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics, 2012, 11(8): 215–229.
[92] Alfaro JF, Gong CX, Monroe ME, et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc Natl Acad Sci USA, 2012, 109(19): 7280–7285.
[93] Minguez P, Letunic I, Parca L, et al. Ptmcode: PTMcode: a database of known and predicted functional associations between post-translational modifications in proteins. Nucleic Acids Res, 2013, 41 (Database issue): D306–311.
(本文责编 陈宏宇)
Progress and application of phosphoproteomics in the proteomics of liver pathological and physiological state
Tailong Yi1,2, Miaomiao Tian2, Xiaoming Yang1,2, and Ping Xu1,2
1 Anhui Medical University, Hefei 230032, Anhui, China 2 Institute of Radiation Medicine, Academy of Military Medical Sciences, Beijing Proteome Research Center, State Key Laboratory of Proteomics, Beijing 102206, China
The phosphorylation is one of most common protein post-translational modifications. The protein phosphorylation plays important roles in the life through the reversible process of phosphorylation and dephosphorylation by kinases and phosphatases. Systematical analysis of the phosphorylation state of proteins would greatly help to reveal the mystery of the life. Recently, with the development of mass spectrometer, bioinformatics sortwares and enrichment methods of phosphopeptides, phosphorylation stduy of orgnism proteins by mass spectrometer has become mature gradually. Liver is one of the most important metabolic and immune organs. In-depth study of protein phosphorylation in liver is of great importance to reveal its function. And booming phosphoproteomics has been applied into the study of liver, which has deepened the knowledge of molecular mechnism of its physiology and pathology states. Here, we review the recent progress on the research and development of phosphoproteomics and their application in liver proteomics study.
proteinphosphorylation, phosphoproteome of liver, enrichment of phosphopeptide, quantitative proteome, bioinformatics
December 27, 2013; Accepted: May 28, 2014
Ping Xu. Tel: +86-10-83147777-1314; Fax: +86-10-80705155; E-mail: xupingghy@gmail.com XiaomingYang. Tel: +86-10-66931424; Fax: +86-10-68212874; E-mail:xiaomingyang@sina.com
衣泰龙, 田苗苗, 杨晓明, 等. 磷酸化蛋白质组学的新进展及其在肝脏生理和病理机制中的应用. 生物工程学报, 2014, 30(7): 1004–1017.
Yi TL, Tian MM, Yang XM, et al. Progress and application of phosphoproteomics in the proteomics of liver pathological and physiological state. Chin J Biotech, 2014, 30(7): 1004–1017.
Supported by: National Basic Research Program of China (973 Program) (Nos. 2011CB910600, 2013CB911200), National High Technology Research and Development Program of China (863 Program) (Nos. SS2012AA020502, 2011AA02A114), National Natural Science Foundation of China (Nos. 31070673, 31170780), Beijing Natural Science Foundation (No. 5112012).
国家重点基础研究发展计划 (973计划) (Nos. 2011CB910600, 2013CB911200),国家高技术研究发展计划 (863计划) (Nos. SS2012AA020502, 2011AA02A114),国家自然科学基金 (Nos. 31070673, 31170780),北京市自然科学基金 (No. 5112012) 资助。
