三氟甲烷下游产品的研究开发
2014-06-05戴佳亮徐卫国金杭丹
戴佳亮 徐卫国 李 华 金杭丹
(浙江省化工研究院有限公司,浙江杭州310023)
三氟甲烷下游产品的研究开发
戴佳亮 徐卫国 李 华 金杭丹
(浙江省化工研究院有限公司,浙江杭州310023)
三氟甲烷在一定条件下会较容易地脱去H,形成活泼的二氟卡宾,可以方便地在医药、农药及特殊化学品中间体内引入三氟甲基和二氟甲基,得到有着特殊功效的含氟化合物。开发三氟甲烷可以节约生产成本,消除环境危害,更可以作为一种新型氟化试剂,在化工、医药、农药展开多种新颖应用。
三氟甲烷;三氟甲基化;二氟甲基化;含氟烃
0 前言
三氟甲烷(HFC-23),CAS号75-46-7,分子式HCF3,相对分子质量70,熔点-160℃,沸点-83℃,相对空气蒸气密度2.43,常态下为无色无臭气体,微溶于水。三氟甲烷主要是HCFC-22(二氟一氯甲烷)及四氟乙烯生产中的副产品,目前可用来制备制冷剂、化学蚀刻剂和清洗剂、复合材料、等离子液体以及氟有机化合物等。三氟甲烷的全球产量为20 000~25 000 t/a。三氟甲烷无毒,不会消耗臭氧层,但是会产生严重的温室效应,在100年内温室效应是CO2的11 700倍,且能在大气层中存在264年[1]。在过去几十年内,三氟甲烷在大气层中的浓度以每年5%的速率稳定增长,造成严重的生态隐患。
处理三氟甲烷有多种方法,如焚烧(氧化)、催化水解、等离子破坏和转化成环境友好物质。本文主要介绍将三氟甲烷转化为环境友好化合物的各种方法,如制备含氟试剂、含氟医药、农药中间体和其余ODS替代品等,减少三氟甲烷的处理成本。
1 制备三氟甲基化合物
目前,有多种药物含有三氟甲基基团,如Fluoxetine(ProzacⓇ)、Celecoxib(CelebrexⓇ)、Mefloquine(LariamⓇ)、Leflunomide(AravaⓇ)、Nilutamide(NilandronⓇ)、Dutasteride(AvodartⓇ)、Bicalutamide(CasodexⓇ)、Aprepitant(EmendⓇ)等。含三氟甲基的农药有Trifluralin、Fipronil、Fluazinam、Penthiopyrad、Picoxystrobin、Fluridone和Norflurazon等[17]。
引入三氟甲基的方法有很多,如自由基取代、亲电亲核取代、加成、卤素置换等。三氟甲烷容易脱去质子形成不稳定的CF-3,分解成二氟卡宾和氟化物,从而可以用来合成各类新型含氟化合物。
1.1 直接三氟甲基化
1991年,日本京都大学的Shono等人[2]首先提出了三氟甲烷可在碱的作用下,去质子化得到三氟甲基阴离子。他们以三氟甲烷与苯甲醛反应为模型来研究各类碱对反应的影响,见式1。DMF溶剂中,NaH为碱时产物2,2,2-三氟甲基-α-苯乙醇收率为28%,叔丁醇钾为碱时产品的收率为40%,而N-烷基取代的吡咯烷酮为碱时产品的收率在34%~80%。在N-正丁基吡咯烷酮/DMF体系,三氟甲基化芳香醛类(如甲氧基苯甲醛、甲基苯甲醛等)均有较好收率60%~90%。而对于酮(芳香酮和烷基酮)类物质,六甲基二硅胺烷(HMDS)则对反应促进效果较好,如三氟甲基化二苯基甲酮,得到2,2,2-三氟甲基-1,1-二苯基乙醇收率84%。1998年,巴黎第十二大学的Troupel等人[3]采用电化学一锅法三氟甲基化芳基醛类物质。在DMF和Bu4NBF4电解液中,以三氟甲烷为甲基化试剂,阴极还原PhI得到强碱,加入芳基醛(苯甲醛、甲基苯甲醛、甲氧基苯甲醛等)为亲电体,5~10℃,通电得到相应的三氟甲基醇类化合物,收率在40%~71%,如式1。

式1
法国巴黎第六大学的Normant等人[4-5]在Shono和Troupel的基础上研究三氟甲烷在金属碱/DMF体系的金属化问题。他们发现在叔丁醇钾存在下,三氟甲烷可得到三氟甲基钾和叔丁醇,但三氟甲基钾在添加金属卤化物后会与叔丁醇转化为叔丁醇盐和三氟甲烷,从而对反应无任何促进效果。于是Normant等人开发了Dimsyl-K碱,来避免叔丁醇对反应的影响,三氟甲基化各类芳基醛如苯甲醛、甲氧基苯甲醛、苯氧基苯甲醛、呋喃甲醛等,见式2,产品分离收率在40%~70%。三氟甲基化苯甲醛得到2,2,2-三氟甲基-α-苯乙醇,收率65%。

式2
法国罗地亚化学公司的Roques等人[6-7]以三氟甲烷/碱/DMF体系三氟甲基化苯甲醛(式3-①)、二苯二硫醚(式3-②)和苯甲酸甲酯(式3-③),得到相应的含三氟甲基化合物,收率均在60%以上。

式3
法国里昂第一大学的Langlois等人[8]设计了一种三氟甲基化试剂1a,其原理为三氟甲烷在DMF中、强碱存在下脱去一个质子形成CF-3基团,它可以在DMF中较稳定地存在,当以N-甲酰基吗啉为强碱时得到1a,可较容易地以硅胶柱层析分离,且收率高(78%)。1a可用于三氟甲基化非烯醇化的羰基化合物,如二苯基甲酮,得到化合物1b,见式4,分离收率可达75%。

式4
2000年,Roques和Langlois等人[9-10]在之前的研究基础上开发了系统性的三氟甲基化体系,主要涉及以三氟甲烷为试剂制备各种含亲电官能团的三氟甲基类化合物,见式5。该体系可三氟甲基化二硫化物(式5-①)、硫氰酸酯(式5-②)、酮类(式5-③)、甲酰胺类(包括甲酰基吗啉)等,产物收率可达60%~90%。

式5
佛罗里达大学的Dolbier等人[11]在2003年的专利中提到了以三氟甲烷和N(SiMe3)3/Me4NF配备的试剂三氟甲基化硫酸酯类化合物,可以得到三氟甲基醇类化合物,如式6。

式6
加州大学的Mukhopadhyay等人[12]在高收率磺化甲烷得到甲磺酸的基础上,研究三氟甲烷的磺化。在强酸性磺化试剂(发烟硫酸、三氧化硫)的作用下,在强碱性溶剂中(t-BuOK和DMF溶液、尿素-H2O2和RhCl3等),三氟甲烷可以转化成三氟甲磺酸,但转化率很低,三氟甲磺酸的选择性更低,均小于10%,且其余产物中六氟乙烷偏多。
2012年,美国南加州大学的Prakash等人[13-14]在多年研究含氟化合物的基础上于Science上发表了系统的直接三氟甲基化的方法。以三氟甲烷在醇盐和金属硅烷盐的存在下,直接三氟甲基化氯硅烷、醛类、酮类、卤代烃、烷基硼酸盐、固体硫等,反应通式见式7。其中制备的三氟甲基(三甲基)硅烷(式7-①),即Ruppert-Prakash试剂Me3SiCF3(TMSCF3),是一种良好的三氟甲基化试剂,但其制备成本高,通常需要以臭氧层消耗物CF3Br为原料,收率可达80%。而三氟甲磺酸也可以式7-③的工艺进行制备。
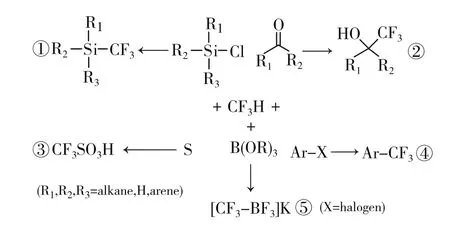
式7
2013年,日本名古屋工业大学的Shibata等人[15]发表了三氟甲烷直接三氟甲基化的研究。他们以2-萘甲醛2a与三氟甲烷为反应模型(见式8),经过选择有机碱(DBU、TMGP4-t-Bu等),溶剂(DMF、THF),探索出适合反应式7-②进行的P4-t-Bu·THF体系,产物2b的收率达到88%。再更换不同羰基化合物为底物,在该体系下,三氟甲基醇类化合物的收率可达50%~92%。
在此基础上,日本东京工业大学的Mikami等人[16]重点研究三氟甲烷直接三氟甲基化反应中有机碱对反应的促进效果。他们同样以式8为反应模型,首先以2-萘甲醛2a和三氟甲烷在溶剂四氢呋喃中,-40℃下反应3 h,探索不同的有机催化剂如TBD、甲基TBD、TBD钾盐、TMG、DBU、P4-t-Bu等对反应的催化作用。只有P4-t-Bu催化后,得到56%的2b,其余催化剂对反应无催化效果。之后以P4-t-Bu为催化剂,筛选此反应的合适溶剂,比较DMSO、DMF、THF、二乙醚、二氧六环等溶剂后,发现在THF中反应效果最好。Mikami等人继续观察不同羰基化合物(醛和酮)在该反应体系(P4-t-Bu,THF,-40℃,3 h)中的反应效果(式7-②),得到的三氟甲基醇类化合物,收率可以在50%~99%之间。更进一步发现,三氟甲烷可在有机碱(P4-t-Bu)作用形成CF-3离子后与酰卤、酯、环氧化合物及二氧化碳等较高效率地反应,其中可与二氧化碳反应得到三氟乙酸,收率达78%。
1.2 制备三氟甲基金属试剂
三氟甲烷是经济、方便、高原子利用率的三氟甲基砌块来源,但是选择性激活H-CF3用于合成三氟甲基化构建砌块和中间体还是有较高难度。通常活化三氟甲烷的方法有经弱酸处理或强碱去质子,产生的CF-3离子很容易降解为二氟卡宾。直到2011年才有文献报道以过渡金属配合物来选择性活化三氟甲烷。
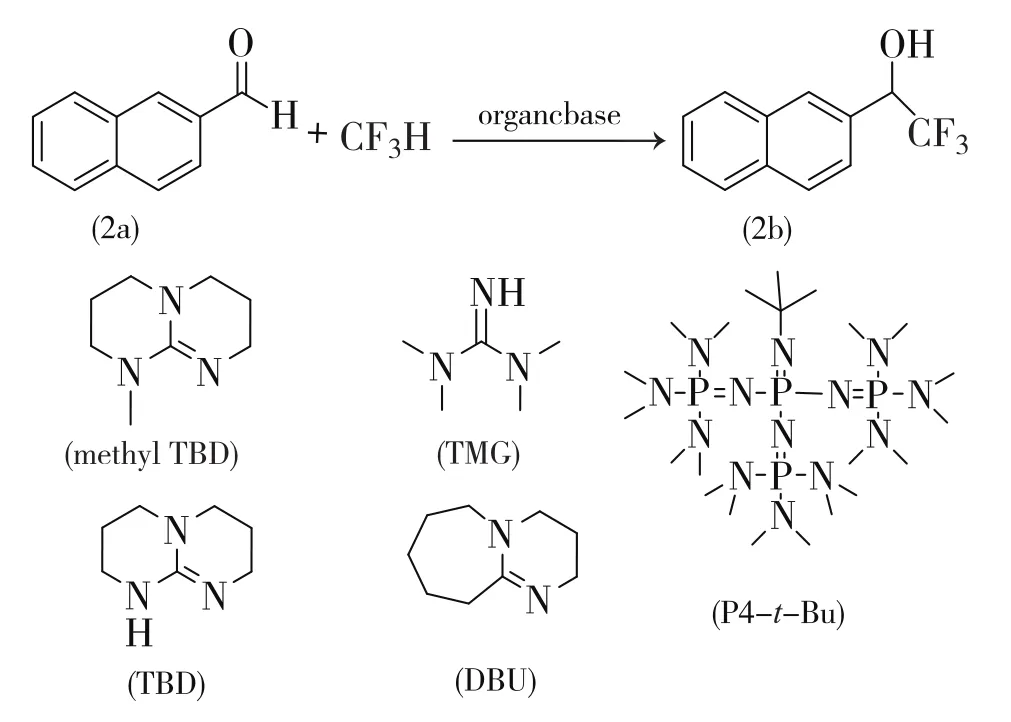
式8
2012年,西班牙加泰罗尼亚化学研究所(ICIQ)的Grushin等人研究开发新的过渡金属配合物活化三氟甲烷体系,并探讨其关键反应机理。首先以三氟甲烷和铜化合物直接制备三氟甲基铜试剂[17-18],从而方便地将活化的三氟甲基导入至其余分子中,得到目标化合物。他们以CuCl为铜试剂,在叔丁醇钾的存在下(Cu∶K=1∶2)制备得到CuCF3,收率可达95%~99%。CuCF3再与碘代或溴代芳烃反应制备得到一系列的三氟甲基芳烃CF3-Ar,如三氟甲苯、对甲基三氟甲苯、对氯三氟甲苯、三氟甲基吡啶、三氟甲基噻吩,19F NMR(核磁共振氟谱)检测CuCF3转化率大于95%,产物收率为75%~99%,工艺见式9-①。
Grushin等人[19]又以三氟甲烷制备得到CuCF3后,亲核三氟甲基化α-卤代芳香酮的C-X键(X为卤素),形成C-CF3键,见式9-②。该反应稳定、快速、条件温和,常温常压下即可反应,且产物收率基本上大于80%。CuCF3试剂中加入Et3N·3HF可延长试剂的活性。

式9
2013年,Grushin等人[20]又以Pd(II)配合物活化三氟甲烷为反应模型,他们发现,在温和的条件下(23℃),有路易斯碱(如n-Bu3P)存在,三氟甲烷可以完全被(dppp)Pd(Ph)(OH)3a活化,形成Pd-CF3键(3b),收率接近100%,反应工艺见式10。其中dppp为1,3-双(二苯基膦)丙烷。

式10
1.3 制备三氟碘甲烷
三氟碘甲烷作为一种有效的三氟甲基合成砌块,还可以替代哈龙1301(CBrF3)和哈龙1211(CBrClF2)作为灭火剂、制冷剂使用,对臭氧危害小,温室效应低。
2002年,日本东曹株式会社的Nagasaki等人[21-23]开发了一种三氟甲烷和碘气相催化制备三氟碘甲烷的方法。首先制备催化剂,探索碱金属催化剂(Rb、Cs、K、Na)的活性,Nagasaki等人发现,铷盐Rb催化下三氟甲烷转化率较高(83%),K盐催化下产物三氟碘甲烷选择性较好(62%)。催化剂载体筛选(SiO2、Al2O3、SiO2-Al2O3、TiO2、活性炭等),选择活性炭时,对反应促进效果最理想,装载KF时三氟甲烷可转化73%。机理研究认为,三氟甲烷于催化剂表面脱去一个HF形成中间态二氟卡宾,且可以催化剂内保持较稳定的状态,再与碘形成三氟碘甲烷。
2006年,美国霍尼韦尔公司的Mukhopadhyay等人[24-25]报道了一步制备三氟碘甲烷的方法,原料为含三氟甲基基团的化合物(如三氟甲烷、三氟溴甲烷、三氟乙酸、三氟乙酰氯、三氟乙酸乙酯等)和碘或其化合物(如一氟化碘、五氟化碘、七氟化碘、碘化氢、氯化碘等)。以三氟甲烷和一氟化碘为原料合成三氟碘甲烷,收率为40%~95%(mol)。氧气的存在对反应影响不大,但可以增加催化剂寿命。
2008年,该公司的Yang等人[26-27]报道了制备碘代氟烃的方法,主要涉及催化剂的制备、处理和再生。碘代氟烃主要是指三氟碘甲烷和五氟碘乙烷,原料为三氟甲烷、五氟乙烷或三氟乙酸等。以三氟甲烷碘化制备三氟碘甲烷为例,反应温度在300~600℃,压力为0.0001~10 MPa,反应保留时间由催化剂床的体积和运料流速比例确定,为0~15 h不等,三氟甲烷的转化率为40%~60%,三氟碘甲烷选择性最高可接近80%,同时还有少量五氟碘乙烷产生。筛选催化剂种类,一般选择以KNO3和La(NO3)3制备的K-La2O3/C且La/K>1[26]。以氮气、三氟甲烷、氢气、碘处理催化剂对反应影响不是很大,但没有氧气存在时,催化剂的活性会明显下降[27]。以一定比例的O2-N2处理催化剂,可活化再生催化剂。
2009年,南京理工大学的潘仁明等人[28-29]采用气相催化法,以三氟甲烷和碘为原料合成了三氟碘甲烷(CF3I)。产物分别经气相质谱、傅里叶变换红外光谱、相对分子质量、催化剂BET比表面积和热重分析等测试,转化率达到52.3%,选择性56.2%,反应后催化剂的比表面积和孔体积降低。
潘仁明与权恒道等人[30]研究了该反应机理,认为其关键点是在高温、催化剂作用下三氟甲烷能够脱HF生成二氟卡宾,二氟卡宾在催化剂表面发生歧化反应生成三氟甲基自由基和炭,三氟甲基自由基与碘反应生成目标物CF3I。催化剂载体选用活性炭可以促进二氟卡宾和三氟甲基自由基的产生。歧化形成的炭沉积在催化剂表面使催化剂BET降低和孔体积减小。
2 制备二氟甲基化合物
二氟甲基官能团能显著增加化合物的亲油性、膜通透性、水溶性和稳定性,被用来合成多种药物,如麻醉剂Desflurane(SupraneⓇ)、降血压药物Riodipine(ForidonⓇ,PhoridoneⓇ,RiosedylⓇ)、抗癌药物Gemcitabine、呼吸系统药物Roflumilast衍生物、抗溃疡(-)-Pantoprazole(RifunⓇ,PantozolⓇ,PantecⓇ)和抗病毒药物二氟甲氧基喹诺酮Garenoxacin[31]。
三氟甲烷可以作为二氟卡宾的来源,制备各类二氟甲基化合物。
2012年,日本东京工业大学的Mikami[32-33]开发了一种制备α-二氟甲基羰基化合物的方法。他们发现羰基化合物的α位可以在碱的作用下脱去质子,与三氟甲烷形成α-二氟甲基羰基化合物,反应通式见式11,收率为20%~70%。反应式见12,产物4b收率为68%。
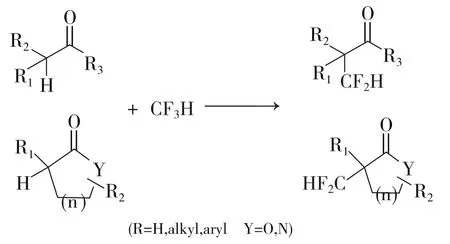
式11
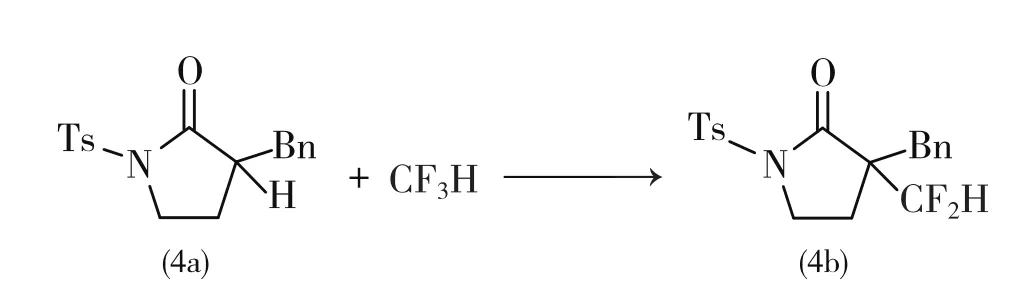
式12
2013年,佛罗里达大学化学系的Thomoson等人[34]由酚类或硫酚类制备芳基二氟甲基醚和二氟甲基硫醚(式13)。该反应为常压、温和的两相反应。首先以对溴苯酚为底物探索反应条件。优选后的反应催化剂为KOH,共溶剂为水,而溶剂为乙腈时,在室温反应效果较好,溶剂为二氧六环时,50℃时反应效果较好。三氟甲烷通过微弱氮气流鼓入反应器中。如三氟甲烷和对溴苯酚反应,以二氧六环为溶剂,得到产物二氟甲氧基对溴苯,经19F NMR(核磁共振氟谱)检测,收率为85%;以乙腈为溶剂,产物二氟甲氧基对溴苯,收率为80%。
Thomoson等人又以多种酚(对甲基苯酚、对氯苯酚、对甲氧基苯酚、萘酚、羟基喹啉等)和硫酚(对甲基、邻甲基硫酚,对氯硫酚等)在该该体系中,选择合适的溶剂乙腈或二氧六环,得到产物二氟甲氧基芳烃或二氟甲硫基芳烃的收率均为60%~90%。
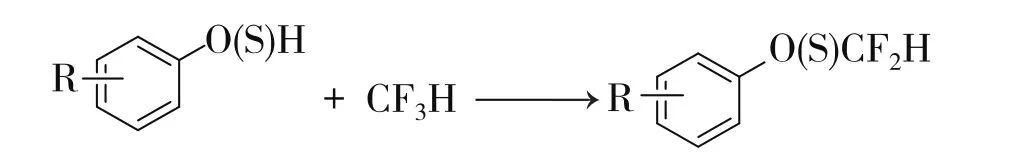
式13
3 制备含氟烷烃
1992年,杜邦公司的Rudershausen[35]将三氟甲烷和四氯化碳进行气相催化反应,歧化重排得到其余各类氟氯甲烷。催化剂选择由Al(NO3)3·9H2O和Mg(NO3)2·6H2O制备的Al/Mg催化剂。三氟甲烷以28 mL/min的速率通入装载有Al/Mg催化剂的反应器中,四氯化碳经蒸发器形成蒸汽后也以同样的速率通入反应器。当反应温度设置为250℃、反应接触时间设为3 s时,产物结果为CCl4(14.5%)、CCl3F(8.4%)、CCl2F2(24.6%)、CHF3(25.4%)、CHCl3(34.1%)和其余少量氟碳化合物。改变温度、原料配比及反应时间后,产物的组成量各有不同。
1997年,杜邦公司的Petrov等人[36-37]由三氟甲烷类物质CF3X和四氟乙烯制备氟代丙烷化合物,催化剂选择铝氯氟化物(ACF,由氯化铝和三氯氟化铝自制)。如以三氟甲烷和四氟乙烯反应可以得到CF3CF2CF2H,精馏得14.6 g CF3CF2CF2H,收率为29.4%,副产物聚四氟乙烯15 g。三氟甲烷也可与三氟乙烯反应得到CF3CFHCF2H,收率只有3%。
2001年,美国佛罗里达大学的Romelaer等人[38]研究三氟甲烷在氢气存在下的高温裂解反应。以铬镍铁合金600为反应器,常压、一定温度下通入三氟甲烷和氢气进行连续反应,三氟甲烷的转化率为21%,产物CF3CH2F、CH2F2和CHF2CHF2的收率分别为39%、26%、5%,碳素平衡82%。经过数据分析,机理可能为三氟甲烷高温生成二氟卡宾CF2∶,其二聚形成四氟乙烯,再由H2中得到H开启自由基链反应得到CF3CH2F、CH2F2和CHF2CHF2。
2002年,昭和电工株式会社的Ohno(大野博基)等人[39-40]公开了一种制备全氟碳化合物的方法,该方法可避免氧化物等杂质的产生,得到高纯度的全氟化碳,可用于半导体工业。其中四氟化碳可以三氟甲烷和氟化氢为原料制备。初步反应四氟化碳含量98.7992%,精馏后再次分析气体成分,四氟化碳含量99.9997%。
2010年,苏威氟化物公司的Bragante等人[41]发明了一种氟化含氯烷烃或氟氯烷烃的方法,主要涉及以三氟甲烷为氟化试剂氟化含氯或氟氯烷烃,如氟化氯仿制备二氟氯甲烷,见式14。以氟化铝为催化剂,氯仿和三氟甲烷经微量泵和流量控制器,以一定比例进入反应器中,反应温度为160~360℃,反应时间由催化剂体积和原料流量的比例来确定。三氟甲烷的转化率最高可达到38%,产物二氟氯甲烷和一氟二氯甲烷的选择性均为4%~8%。

式14
2013年,常熟三爱富中昊化工新材料有限公司的司林旭等人[42]开发了一种三氟甲烷裂解制备二氟一氯甲烷的工艺。将原料三氟甲烷、甲烷氯化物按照物质的量比0.1~10混合投入填充催化剂的反应器,在150~350℃的温度下保留3~30 s进行裂解反应,得到含有三氟甲烷、甲烷氯化物、一氟二氯甲烷和二氟一氯甲烷的混合物,二氟一氯甲烷直接通过分离获得;副产物一氟二氯甲烷与氟化氢再反应生成二氟一氯甲烷;混合物中的三氟甲烷、甲烷氯化物分离回收后继续作为反应混合物。该方法大幅避免了三氟甲烷排放对环境造成的危害,三氟甲烷转化率在40%以上,产物(包含为反应原料)中二氟一氯甲烷的含量可在20%以上。
4 制备含氟烯烃
氢氟烯烃(HFO),如氟代丙烯等相对于氢氟碳化合物(HFC)而言,预期对臭氧气层影响小,GWP较低,可燃性和毒性低。
2002年,韩国科学技术院的Moon等人[43-45]以三氟甲烷和四氟乙烯(TFE)混合裂解制备六氟丙烯(HFP)。反应在氮气保护下于铬镍铁合金600管式反应器内进行,优选的条件为HFC-23/TFE为1~4,裂解温度在850~900℃,接触反应时间为0.5~2 s。反应后主要的副产物有全氟异丁烯(CF3)2C=CF2(PFIB)、CF3C=CCF3、C2F3H等。其由三氟甲烷生成六氟丙烯的连锁反应机理可能为三氟甲烷的裂解和四氟乙烯的二聚及三氟甲烷裂解产生的中间态物质的相互作用,顺序为HFC-23→TFE→C4F8→HFP→PFIB,其中最多的有毒副产物全氟异丁烯PFIB可能是由六氟丙烯和二氟卡宾[∶CF2]形成的。控制反应温度(维持热量平衡)和接触反应时间可得到高纯度的六氟丙烯,并减少PFIB、聚四氟乙烯和积炭的产生。如将水蒸气和三氟甲烷混合后裂解,则主要产物为CO2、CO、H2和CF4。
2003年,杜邦公司的Rao等人[46]将三氟甲烷和二氟氯甲烷(HCFC-22)共同热解,在690~775℃,接触时间1~2 s,可以得到四氟乙烯、五氟乙烷(HFC-125)、六氟丙烯等其余氟碳产品。调整三氟甲烷和二氟氯甲烷的比例和反应温度,四氟乙烯在产物中的含量可达25%左右,HFC-125含量在3%左右,HFP可至8%。单独热解三氟甲烷,转化率基本上小于5%。
2004年,Iikubo等人[47]开发了一种将氢氟烃(HFC-152、HFC-32、HFC-23等)转化为含氟烯烃(三氟丙烯、四氟乙烯、二氟乙烯等)的方法。如可以将HFC-23(三氟甲烷)先转化为CFC-13(三氟氯甲烷),再得到三氟丙烯(TFP),见式15。
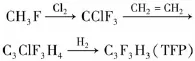
式15
2010年,澳大利亚纽卡斯尔大学的Kennedy和韩文锋等人[48]用三氟甲烷和甲烷气相反应得到偏二氟乙烯,并研究了各类催化剂对反应的促进作用[49-53]。偏二氟乙烯(CH2=CF2)作为含氟单体,可用来合成各种氟材料,如PVDF(聚偏二氟乙烯)、Viton(杜邦公司生产)、KEL-F(3M)和Aflas(旭硝子)氟橡胶。
在400~900℃下,三氟甲烷和甲烷直接气相反应会生成大量的副产物四氟乙烯,添加CaBr2能促进三氟甲烷和甲烷的转化,且基本上不产生四氟乙烯[49]。探讨机理认为,三氟甲烷和CaBr2经过交互作用,形成CaF2并释放活性Br,得到CBrF3。随后Br、CBrF3、甲烷三者作用生成偏二氟乙烯。
在少量甲醇(5%的三氟甲烷浓度)的存在下[50],温度900~1 150 K,在氧化铝反应管中反应,三氟甲烷和甲烷的转化率可增加50%~100%,偏二氟乙烯的生成速率也相应增加。当甲醇含量增加至50%时,在873~1 123 K下反应,三氟甲烷转化率可增加,但偏二氟乙烯的形成速率并未改变。
在痕量的CBrF3存在下[51],温度873 K和1 173 K,三氟甲烷和甲烷分解后可以得到主要产品偏二氟乙烯,副产品乙烯、二氟甲烷、四氟乙烯等。实验验证,6 000×10-6的CBrF3能明显增加甲烷的转化率,从而使偏二氟乙烯含量增大,副产品量降低。但增加CBrF3对三氟甲烷转化率不产生影响。
经过验证,甲醇和CBrF3同时存在,对三氟甲烷的分解有促进作用[52]。
在没有甲烷存在下,以负载KNO3的活性炭为催化剂[53],温度873~1 173 K、空速4 300 h-1下分解三氟甲烷,反应活性较高且相对稳定,三氟甲烷的转化率相对于直接气相反应提高了10倍,主要产物偏二氟乙烯和六氟丙烯的选择性分别为55%和35%,产生的HF则与钾形成了KF。
2011年,陶氏环球技术有限责任公司的Tirtowidjojo等人[54]开发了一种生产氯代或氟代丙烯的有效方法。该方法产物收率较好,残余物和副产物浓度低(低于20%甚至10%),且可在低于500℃下进行,节约能量。以三氟甲烷和三氯乙烯生产1,1-二氯-3,3,3-三氟丙烯为例,转化率为12%左右,产品选择性大于50%。
5 直接裂解
1999年,Rossin等人[55]以ZrO2和ZrO2-SO4为催化剂,在水的存在下,通过固定床催化降解三氟甲烷。该反应原理为催化水解反应,加入硫酸能增加催化剂的活性、降低降解温度至40℃。水作为降解三氟甲烷的原料,还能增强催化剂的稳定性。三氟甲烷降解后的产物为CO、HF和CO2,而残余的氟则留在催化剂中,降低催化剂表面积甚至导致催化剂失活。
2012年,斯洛文尼亚Jozef Stefan Institute(JSI)的Skapin等人[56]研究了在温和条件下分解三氟甲烷的工艺。三氟甲烷与固体氢氧化物如氢氧化钾、氢氧化钠、氢氧化锂等,在温度370~484 K下分解;与碳酸盐如碳酸钠、碳酸钾、碳酸锂等,分解温度要高100~150 K,分解产生CO、水、氟化金属盐,CO和水可通过水煤气反应转化为碳酸盐。Skapin等人认为,分解温度可能与碱性强弱有关,如氢氧化钠、氢氧化钾可在较低的温度(420~470 K)下完全分解三氟甲烷,酸碱相互作用对此分解反应的进行起着重要的作用。
6 其他
2008年,日本大金公司的Takubo等人[57]公开了一种制备羰基氟的方法,以三氟甲烷和氧气为原料制备得到碳酰氟和副产物氟化氢、二氧化碳等,优选反应温度300~1 000℃,反应时间0~30 min,氧气相对三氟甲烷比例为0.5~50∶1,反应式见式16。红外光谱分析三氟甲烷转化率为99%,碳酰氟选择性为99.8%。
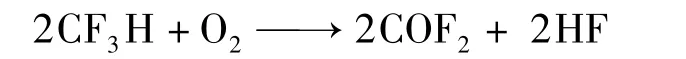
式16
2012年,加拿大EPIC企业的Berrang[58]开发了一项制备Poly(hydridocarbyne)聚碳炔氢(PHC)的工艺,PHC可用来制备金刚石、高硬度的类钻碳膜、胶黏剂等。三氟甲烷可作为制备PHC的一种原料(式17)。以镁或锌为电极,首先在-82℃下将三氟甲烷转化为液相,避免了其余种类化合物(主要是溶剂)的引入。由于三氟甲烷在反应中活性不高,可以加入MgF2或ZnF2、无机酸(HF)等促进液相的电导率。反应在惰性气体保护下进行,通入合适的微弱电流防止三氟甲烷率先分解,完成反应后,升高温度至室温,三氟甲烷气化,MgF2沉淀,PHC经萃取分离,可得到高纯度、高收率的PHC产品。

式17
7 结论
三氟甲烷作为生产四氟乙烯的副产品,在大气层中的浓度逐年递增,将会造成严重的生态隐患。但三氟甲烷在一定条件下会较容易地脱去H,形成活泼的二氟卡宾,可以方便地在医药和农药中间体内引入三氟甲基和二氟甲基,得到有着特殊功效的含氟化合物;还可以将其转化为各类氟氯烷烃和烯烃及对环境友好的新型ODS替代品。
综上所述,对三氟甲烷的进一步应用研究,可以节约生产成本,消除环境危害,更可以作为一种新型氟化试剂,在化工、医药、农药展开多种新颖应用。
[1]Han W,Li Y,Tang H,et al.Treatment of the potent greenhouse gas[J],CHF3-An overview[J].J.Fluorine Chem.,2012,140:7-16.
[2]Shono T,Ishifune M,Okada T,et al.Electroorganic chemistrys[J].130.A novel trifluoromethylation of aldehydes and ketones promoted by an electrogenerated base[J].J.Org.Chem.,1991,56:2-4.
[3]Barhdadi R,Troupel M,Perichon J.et al.Coupling of fluoroform with aldehydes using an electrogenerated base[J].Chem.Commun.,1998(12):1251-1252.
[4]Folleas B,Marek I,Normant J F,et al.Fluoroform:an efficient precursor for the trifluoromethylation of aldehydes[J].Tetrahedron Letters,1998,39(19):2973-2976.
[5]Folleas B,Marek I,Normant J F,et al.Fluoroform:an efficient precursor for the trifluoromethylation of aldehydes[J].Tetrahedron,2000,56(2):275-283.
[6]Russell J,Roques N.Effective nucleophilic trifluoromethylation with fluoroform and common base[J].Tetrahedron,1998,54(45):13771-13782.
[7]Roques N,Russell J.Perfluoroalkylation method and reagent therefor:US,6096926[P].2000-08-01.
[8]Billard T,Bruns S,Langlois B R.New stable reagents for the nucleophilic trifluoromethylation.1.Trifluoromethylation of carbonyl compoundswith N-formylmorpholine derivatives[J].Org.Lett.,2000,2(14):2101-2103.
[9]Roques N,Russell J,Langlois B,et al.Compounds useful for perhalogenoalkylation,reagent for implementing these compounds and synthesis method for obtaining these compounds:US,6203721[P].2001-03-20.
[10]Large S,Roques N,Langlois BR,etal.Nucleophilic trifluoromethylation of carbonyl compounds and disulfideswithtrifluoromethane and silicon-containing bases[J].J.Org.Chem.,2000,65(26):8848-8856.
[11]Dolbier W R,Takechi N.Process for trifluoromethylation of sulfates:WO,2003078366[P].2003-09-25.
[12]Mukhopadhyay S,Bell A T,Srinivas R V.etal.Synthesis of trifluoromethanesulfonic acid from CHF3[J].Org.Process Res.Dev.,2004,8(4):660-662.
[13]Prakash G K Surya,Jog PV,Batamack Patrice TD,et al.Direct trifluoromethylations using trifluoromethane:WO,2012148772[P].2012-11-01.
[14]Prakash G K,Jog PV,Batamack P TD,et al.Taming of fluoroform:direct nucleophilic trifluoromethylation of Si,B,S,and C centers[J].Science,2012,338(6112):1324-1327.
[15]Kawai H,Yuan Z,Shibata N,et al.A sterically demanding organo-superbase avoids decomposition of a naked trifluoromethyl carbanion directly generated from fluoroform[J].Org.Biomol.Chem.,2013,11(9):1446-1450.
[16]Zhang Y,Fujiu M,Mikami K,et al.Organocatalysis approach to trifluoromethylation with fluoroform[J].J.Fluorine Chem.,2013,156:367-371.
[17]Grushin V,Zanardi A.Process to obtain a trifluoromethylating composition:WO,2012113726[P].2012-08-30.
[18]Zanardi A,Novikov M A,Grushin V V,et al.Direct cupration of fluoroform[J].J.Am.Chem.Soc.,2011,133(51):20901-20913.
[19]Novak P,Lishchynskyi A,Grushin V V,et al.Trifluoromethylation ofα-haloketones[J].J.Am.Chem.Soc.,2012,134(39):16167-16170.
[20]Takemoto S,Grushin V V.Nucleophile-catalyzed,facile,and highly selective C-H activation of fluoroform with Pd(II)[J].J.Am.Chem.Soc.,2013,135(45):16837-16840.
[21]Nagasaki N,Suzuki N,Arai S,et al.The development of a novel catalytic technology for CF3I manufacture[J].Speciality Chemicals Magazine,2002,22(5):31-32.
[22]Nagasaki N Morikuni Y,Kawada K,et al.Study on a novel catalytic reaction and itsmechanism for CF3Isynthesis[J].Catal.Today,2004,88:121-126.
[23]Nagasaki N,Morikuni Y,Kawada K,et al.Method for producing trifluoroiodomethane and installation therefor:JP,2005008543[P].2005-01-13.
[24]Mukhopadhyay S,Tung H S.One-step synthesis of CF3I:US,7132578[P].2006-11-07.
[25]Mukhopadhyay S,Tung H S.Direct one-step synthesis from CF3-I:WO,2006063241[P].2006-06-15.
[26]Yang S,Tung H S.Catalyst for the synthesis of CF3I and CF3CF2I:US,20080200735[P].2008-08-21.
[27]Yang S,Tung H S.Method for pretreating and regenerating catalysts used in a process for making fluoroiodoalkanes:US,20090137852[P].2009-05-28.
[28]李勤华,谈玲华,潘仁明,等.三氟碘甲烷的合成及机理[J].南京理工大学学报,2011,35(6):863-866.
[29]杨光成,冒爱勤,潘仁明,等.三氟碘甲烷气相合成催化剂分析[J].工业催化,2009,17(11):66-69.
[30]Yang G C,Pan R M,Quan H D,et al.Investigation of CF2carbene on the surface of activated charcoal in the synthesis of trifluoroiodomethane via vapor-phase catalytic reaction[J].J.Fluorine Chem.,2009,130(2):231-235.
[31]Jeschke P,Baston E,Leroux,FR,et al.α-Fluorinated ethers as"exotic"entity in medicinal chemistry[J].Mini-Rev.Med.Chem.,2007,107:1027-1034.
[32]Mikami K.Method for producingα-fluoromethylcarbonyl compound:JP,2012051825[P].2012-03-15.
[33]Iida T,Hashimoto R,Mikami K.et al.Umpolung of fluoroform by C-F bond activation:direct difluoromethylation of lithium enolates[J].Angew.Chem.Int.Ed.,2012,51(38):9535-9538.
[34]Thomoson CS,DolbierW R.Use of fluoroform as a source of difluorocarbene in the synthesis of difluoromethoxy and difluorothiomethoxyarenes[J].J.Org.Chem.,2013,78(17):8904-8908.
[35]Rudershausen C G.Disproportionation of selected chlorofluoromethanes:US,5146020[P].1992-09-08.
[36]Petrov V A,Krespan C G.Addition of some unreactive fluoroalkanes to tetrafluoroethylene.Direct catalytic synthesis of F-butene-2[J].J.Fluorine Chem.,2000,102:199-204.
[37]Krespan CG,Petrov V A.Addition of trifluoromethanes to fluoroolefins and isomerization of monohaloperfluoro alkanes:WO,9702227[P].1997-01-23.
[38]Romelaer R,Kruger V,Baker JM,et al.Pyrolyses of chlorodifluoromethane and trifluoromethane in the presence of hydrogen.Mechanism and optimization of reaction conditions[J].J.Am.Chem.Soc.,2001,123(28):6767-6772.
[39]Ohno H,Ohi T.Method for producing perfluorocarbons and application thereof:JP,2002255868[P].2002-09-11.
[40]Ohno H,Ohi T.Process for producing perfluorocarbons and use thereof:WO,2002066408[P].2002-08-29.
[41]Bragante L,Peron S,Unveren E,et al.Process for the manufacture of hydrochlorofluorocarbons using trifluoromethane as fluorinating agent:EP,2172441[P].2010-04-07.
[42]司林旭,王寅洁,沈达,等.一种三氟甲烷裂解制备二氟一氯甲烷的工艺方法:中国,103467239[P].2013-12-25.
[43]Moon D J,Ahn Byoung S.Pyrolysis of a mixture of trifluoromethane and tetrafluoroethylene to produce hexafluoropropylene[J].J.Chem.Eng.Jpn.,2004,37(2):318-325.
[44]Moon D J,Chung M J,Kin H G,etal.Pyrolysis of trifluoromethane to produce hexafluoropropylene[J].Ind.Eng.Chem.Res.,2002,41(12):2895-2902.
[45]Moon D J,Kim H G,Byoung S A.et al.Preparation of hexafluoropropylene from the pyrolysis of trifluoromethane and tetrafluoroethylene:US,2002087038[P].2002-07-04.
[46]Rao V N M,Gelblum PG,Noelke C J,et al.Disposal of fluoroform(HFC-23):WO,2003051802[P].2003-06-26.
[47]Iikubo Y,Hedrick V,Brandstadter SM,et al.Materials and methods for the conversion of hydrofluorocarbons:US,20040127757[P].2004-07-01.
[48]Han W,Kennedy E M,Kundu SK,et al.Experimental and chemical kinetic study of the pyrolysis of trifluoroethane and the reaction of trifluoromethane with methane[J].J.Fluorine Chem.,2010,131(7):751-760.
[49]Han W,Yu H,Kennedy E M,et al.Conversion of CHF3to CH2=CF2via reaction with CH4and CaBr2[J].Environ.Sci.Technol.,2008,42(15):5795-5799.
[50]Han W,Kennedy EM,Mackie JC,etal.Effectofmethanol on the gas-phase reaction of trifluoromethane with methane[J].Ind.Eng.Chem.Res.,2010,49(18):8406-8414.
[51]Han W,Kennedy E M.Mackie JC,et al.Conversion of CHF3to CH2=CF2via reaction with CH4in the presence of CBrF3:An experimental and kinetic modelling study[J].J.Hazard.Mater.,2010,180:181-187.
[52]Han W,Kennedy E M,Mackie J C,et al.Mechanistic study of the reaction of CHF3with CH4[J].Chem.Eng.J.,2011,166(3):822-831.
[53]Han W,Kennedy EM,Liu H,etal.Catalytic pyrolysis of CHF3over activated carbon and activated carbon supported potassium catalyst[J].J.Fluorine Chem.,2010,131(6):698-703.
[54]Tirtowidjojo M M,Chakraborty D,Eiffler J,etal.Process for the production of chlorinated and/or fluorinated propenes:WO,2011044447[P].2011-04-14.
[55]Feaver W B,Rossin J A.Interaction of trifluoromethane(CHF3)with alkalihydroxides and carbonates[J].Catal.Today,1999,54(1):13-22.
[56]Vakulka A,Tavcar G,Skapin T,et al.Interaction of trifluoromethane(CHF3)with alkali hydroxides and carbonates[J].J.Fluorine Chem.,2012,142:52-59.
[57]Takubo S,Kume T,Yamamoto A,et al.Method for producing carbonyl difluoride:US,2008021243[P].2008-01-24.
[58]Berrang P G.Method for making poly(hydridocarbyne):WO,2012103622[P].2012-08-09.
The Research and Development for Downstream Products of Trifluoromethane
Dai Jialiang,Xu Weiguo,Li Hua,Jin Hangdan
(Zhejiang Chemical Industry Research Institute Co.,Ltd.,Hangzhou 310023,China)
Trifluoromethane under certain conditions will be form difluorocarbene by remove a H,activation for the synthesis of trifluoromethylated or difluoromethylated intermediates,obtain valuable fuctionalized organic compounds.Available to treat trifluoromethane,itwould be economically and environmentally more sustainable to feed stocks for production of novel fluoride applications in pharmaceutical,agrochemical and specialty materials industries.
trifluoromethane;trifluoromethylation;difluoromethylation;hydrofluorocarbon
戴佳亮(1984—),男,工程师。主要从事含氟精细化学品的研究开发工作。
