国内外标准测定依达拉奉注射液含量结果的对比研究
2014-06-01孙莹林大专孙全乐张凌赢
孙莹 林大专 孙全乐 张凌赢
依达拉奉注射液是日本三菱东京制药株式会社开发的中枢神经系统用药, 于2001年在日本首次上市, 随后国内多家企业相继上市仿制药。在质量标准的含量测定项下, 国内外标准均采用高效液相色谱法, 但日本标准采用的是内标法,国内标准采用的是外标法, 为了比较国内外标准测定依达拉奉注射液中依达拉奉含量结果的一致性, 我们按照日本标准和国内标准分别对同一厂家的三批依达拉奉注射液中依达拉奉的含量进行了测定, 以其确定国内标准的可行性。
1 仪器与试药
1.1 主要仪器 Agilent 1200高效液相色谱仪(Agilent 1200可变波长检测器(VWD), Agilent 1200数据站, P/N-7725i手动进样器, G1354A四元梯度泵及G1316A柱温箱(美国Agilent公司产品)。
1.2 主要试药 依达拉奉对照品(南通飞宇生物科技有限公司, 批号FY24810511, 规格:100mg, 含量≥99%);依达拉奉注射液(吉林省某药业股份有限公司:2013063001、2013063002、2013063003);甲醇 (美国 Fisher公司 , 色谱纯 );醋酸铵(北京化工厂, 分析纯);乙腈(美国Fisher公司, 色谱纯);醋酸(北京化工厂, 分析纯);氨水(北京化工厂, 分析纯);对氨基苯甲酸乙酯(阿拉丁化学试剂上海有限公司,分析纯)。
2 方法与结果
2.1 色谱条件
2.1.1 国内标准[1]Kromasil5~C18 柱 (250 mm×4.6 mm,5 μm);流动相为0.05 mol/L醋酸铵溶液(用冰醋酸调节pH=4.0±0.1)-乙腈(75:25);流速1.0 ml/min;柱温25℃;检测波长 254 nm ;进样量 20 μl。
对照品溶液的制备:取依达拉奉对照品适量, 精密称定,加流动相溶解并稀释制成每毫升中约含依达拉奉20 μg的溶液。
供试品溶液的制备:精密量取依达拉奉注射液0.70 ml,加流动相稀释至50 ml。
2.1.2 日本标准[2]Kromasil5-C18 柱 (150 mm×4.6 mm,5 μm);流动相为稀乙酸(1→100)-甲醇(3:1)加入氨试液调节pH至5.5;流速1.5 ml/min;柱温50℃;检测波长240 nm ;进样量 20 μl。
对照品溶液的制备:取依达拉奉对照品适量, 精密称定,加内标溶液2.50 ml, 用甲醇稀释至50 ml。
供试品溶液的制备:精密量取依达拉奉注射液0.50 ml,加内标溶液2.50 ml, 用甲醇稀释至50 ml。
内标溶液的制备:精密称定氨基苯甲酸乙酯约0.5 g, 用甲醇稀释至250 ml。
2.1.3 系统适用性试验 在上述色谱条件下, 分别精密量取依达拉奉对照品和供试品溶液各20 μl注入液相色谱仪, 理论板数以依达拉奉峰计算分别为2552(国内标准)和5436(日本标准 )。
2.2 含量测定方法
2.2.1 国内标准 精密量取依达拉奉对照品及供试品溶液各20μl注入液相色谱仪, 记录色谱图, 按外标法以峰面积计算依达拉奉注射液中依达拉奉的含量。
2.2.2 日本标准 取本品适量, 约相当于依达拉奉3 mg, 精密加入对氨基苯甲酸乙酯溶液10 ml, 加甲醇稀释至20 ml, 作为供试品溶液;取依达拉奉对照品75 mg, 精密称定, 用甲醇定量稀释至50 ml量瓶中, 精密量取2 ml, 精密加入内标溶液10 ml, 加甲醇稀释至20 ml, 作为对照品溶液。取供试品和对照品溶液各2 μl, 注入液相色谱仪, 以峰面积计算。
2.3 结果
2.3.1 国内标准
2.3.1.1 线性试验 精密称定干燥至恒重的依达拉奉对照品适量, 加流动相制成990 μg/ml的贮备液, 分别精密量取上述贮备液 0.6、0.8、1.0、1.2、1.4 ml置 50 ml容量瓶中, 加流动相稀释至刻度。精密量取上述溶液20 μl, 注入液相色谱仪, 记录色谱图及峰面积。以溶液浓度(X)为横坐标, 相应峰面积(Y)为纵坐标进行线性回归, 得回归方程Y=52.07x+50.59(r=0.9992)。依达拉奉在 11.88~27.72 μg/ml范围内, 浓度与峰面积呈良好的线性关系。
2.3.1.2 精密度试验 精密量取对照品溶液各20 μl, 连续进样6次, 记录各次测定的依达拉奉峰面积, 峰面积RSD值为0.74%。
2.3.1.3 稳定性试验 取同一份供试品溶液, 分别于0、2、4、8、12 h依法测定, 峰面积RSD值为0.6%, 供试品溶液在12 h内稳定。
2.3.1.4 重复性试验 精密量取批号为2013063001样品6份, 按供试品溶液制备方法制备, 依法测定, 结果六份样品溶液的平均含量为96.8%, RSD值为0.7%。
2.3.1.5 回收率试验 精密量取已知含量的供试品溶液9份, 每份0.6 ml, 置50 ml量瓶中, 分别精密加入对照品溶液(19.8 μg/ml)4.5、9.5、14.5 ml, 制成高、中、低三个浓度的溶液 ,各3份, 依法测定, 计算回收率, 平均回收率为100.3%, RSD为0.462%。
2.3.1.6 样品测定 取本品三批样品依法测定, 平均含量95.0%(占标示量的100%)RSD为0.122%。
2.3.2 日本标准
2.3.2.1 线性试验 精密称定干燥至恒重的依达拉奉对照品适量, 加流动相制成990 μg/ml的贮备液, 分别精密量取上述贮备液0.56、0.65、0.46、0.85、0.95 ml置50 ml容量瓶中,分别加对氨基苯甲酸乙酯溶液2.5 ml, 加甲醇稀释至刻度。精密量取上述溶液20 μl, 注入液相色谱仪, 记录色谱图及峰面积。以溶液浓度(X)为横坐标, 相应依达拉奉峰面积(Y)为纵坐标进行线性回归, 得回归方程Y=48.120x+14.080(r=0.9994)。依达拉奉在11.088~18.81μg/ml范围内, 浓度与峰面积呈良好的线性关系。
2.3.2.2 精密度试验 精密量取对照品溶液(含内标液)各20 μl, 连续进样6次, 记录各次测定的依达拉奉峰面积, 峰面积RSD值为0.95%。
2.3.2.3 稳定性试验 取同一份供试品溶液(含内标液),分别于0、2、4、8、12 h依法测定, 依达拉奉峰峰面积RSD值为0.7%, 供试品溶液在12 h内稳定。
2.3.2.4 重复性试验 精密量取批号为2013063001样品6份, 按2.1.2日本标准供试品溶液制备方法制备, 依法测定,结果六份样品溶液的平均含量为98.5%, RSD值为0.327%。
2.3.2.5 回收率试验 精密量取已知含量的供试品溶液9份, 每份0.1 ml, 置50 ml量瓶中, 分别精密加入对照品溶液(990 μg/ml)0.5、0.6、0.7 ml, 制成高、中、低三个浓度的溶液,各3份, 再分别精密加入内标溶液(1→500)2.5 ml, 依法测定,计算回收率, 平均回收率为100.2%, RSD为0.526%。
2.3.2.6 样品测定 取本品三批样品依法测定, 平均含量(占标示量的100%)为98.2%, RSD为0.305%,
2.3.3 结果分析与讨论
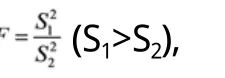
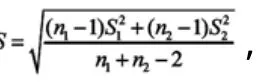
3 讨论
本项研究依据文献比较了国内外标准测定含量结果的一致性, 通过F检验和t检验, 得出两种方法的精密度和测定的依达拉奉含量平均值之间没有显著性差异, 说明作为含量测定方法二者可以互相代替, 尤其是日本标准(内标法)操作比较繁琐、费时耗材高, 所以国内标准(外标法)完全可以替代日本原标准用于依达拉奉注射液中依达拉奉含量的控制。
[1] 国家食品药品监督管理局.依达拉奉注射液标准(试行).YBH03182005.
[2] 厚生省.日本药局方.第 11 改正解说书 (一部) , 1986:56.
[3] 古铁波, 陈贵平.高效液相色谱法测定依达拉奉注射液中依达拉奉及有关物质的含量.中国当代医药, 2013, 20(21):61-65.
[4] 齐莹.HPLC法测定依达拉奉注射液的含量.黑龙江科技信息,2013(3):19.
