RP-HPLC法测定康普瑞丁A4磷酸酯聚合物囊泡的含量
2014-05-22朱金芳胡梦莹邱利焱新疆农业大学食品科学与药学学院乌鲁木齐83005浙江大学药学院杭州310058
朱金芳,胡梦莹,邱利焱#(1.新疆农业大学食品科学与药学学院,乌鲁木齐 83005;.浙江大学药学院,杭州 310058)
从南非灌木Combretum caffrum中分离得到的康普瑞丁A4(CA4)是一种作用于秋水仙碱位点的微管蛋白结合剂,康普瑞丁A4磷酸酯(CA4P)是CA4的水溶性的磷酸酯化前体药物,是第1个低于最大耐受量(MTD)就可以选择性地产生抗肿瘤血管作用的小分子血管破坏药物[1]。CA4P不仅可特异性作用于肿瘤血管系统而不影响正常组织,快速破坏实体肿瘤血管,阻断肿瘤血液供应,而且具有抑制癌细胞增长及放化疗增敏的作用[2],该药在国外已进入Ⅲ期临床研究阶段。CA4P注射剂对光和热不稳定,在动物体内半衰期短,在体内消除很快[3-4]。鉴于此,笔者将其包封于生物可降解的高分子聚合物囊泡中,增加了药物的稳定性,使CA4P缓慢持续释放,延长其在体内的作用时间,减少给药频率;并能通过纳米囊泡的肿瘤组织增强渗透滞留作用(EPR effect)使CA4P被动靶向于肿瘤组织,从而提高疗效、降低毒副作用。本文采用反相高效液相色谱法(RP-HPLC)测定CA4P聚合物/囊泡中CA4P的含量,系统地考察了方法的准确度、精密度及稳定性等,以期为CA4P聚合物/囊泡的质量控制提供依据。
1 材料
1.1 仪器
AL104精密电子天平(瑞士梅特勒-托利多仪器有限公司);1100 HPLC仪(美国Agilent公司);TU-1800PC/SPC紫外分光光度计(北京普析通用仪器有限责任公司)。
1.2 药品与试剂
CA4P对照品(四川大学提供,批号:20110310,纯度:99.6%);CA4P聚合物/囊泡(浙江大学药学院药物制剂研究所,批号:20120718、20120719、20120720,含量:含CA4P 190 μg/ml);乙腈、甲醇为色谱纯,水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Diamonsil C18(150 mm×4.6 mm,5 μm);流动相:乙腈-甲醇-0.02 mol/L乙酸铵(10∶40∶50,pH为6.6),流速:1.0 ml/min;检测波长:288 nm;进样量:20 μl。
2.2 溶液的制备
对照品贮备液:精密称取CA4P对照品适量,用流动相配制成含CA4P 210 μg/ml的溶液,摇匀即得。
对照品溶液:精密量取CA4P对照品贮备液适量,用流动相稀释成含CA4P 10.5 μg/ml的溶液,摇匀,用0.22 μm的微孔滤膜过滤,取续滤液,即得。
供试品溶液:精密量取CA4P聚合物/囊泡溶液400 μl置于5 ml棕色量瓶中,用1 ml乙腈破坏囊泡,使药物从囊泡中释放出来,用流动相定容至刻度,摇匀,用0.22 μm的微孔滤膜过滤,取续滤液,即得。
阴性对照溶液:取不加CA4P的空白囊泡溶液400 μl置于5 ml棕色量瓶中,用1 ml乙腈破坏囊泡,用流动相定容至刻度,摇匀,用0.22 μm的微孔滤膜过滤,取续滤液,即得。
2.3 检测波长的选择
取上述对照品溶液,在600~190 nm波长范围内进行波长扫描,确定最大吸收波长。结果显示,对照品溶液在288 nm波长处左右有最大吸收,故选定288 nm为测定波长。
2.4 系统适用性试验
在“2.1”项色谱条件下,取空白溶液(溶剂)、阴性对照溶液、对照品溶液和供试品溶液(批号:20120718)进样,结果显示,理论板数按CA4P峰计算>3000,供试品中CA4P峰与相近峰分离清晰完全,分离度>1.5,空白和阴性对照溶液对CA4P主峰未见干扰,见图1。
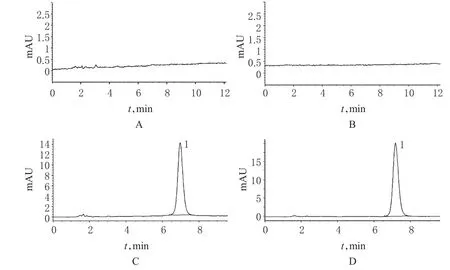
图1 系统适用性试验高效液相色谱图A.空白;B.阴性对照;C.对照品;D.供试品;1.CA4PFig 1 HPLC chromatograms of system suitability testA.blank;B.negative control;C.reference substance;D.test sample;1.CA4P
2.5 线性关系考察
精密量取CA4P对照品贮备液(210 μg/ml)100、200、500 μl及1、2 ml,分别置于10 ml棕色量瓶中,用流动相定容至刻度,摇匀,用0.22 μm的微孔滤膜过滤。分别精密吸取续滤液20 μl注入色谱仪,记录色谱图。以CA4P的峰面积(y)为纵坐标、CA4P的质量浓度(x)为横坐标,进行线性回归,得回归方程为y=29.651x-9.6083(r=0.9999)。结果表明,CA4P检测质量浓度线性范围为2.1~42 μg/ml。
2.6 检测限与定量限试验
以CA4P的色谱峰信噪比为3时的质量浓度为检测限,信噪比为10时的质量浓度为定量限。取标准曲线最低质量浓度(2.10 μg/ml)对照品溶液稀释10倍后进样检测,其信噪比>3,因此检测限定为0.21 μg/ml;稀释3倍后进样检测,其信噪比>10,因此定量限定为0.70 μg/ml,结果见图2。
2.7 精密度试验
取CA4P对照品溶液,分别注入色谱仪测定5次,记录峰面积,计算其RSD=1.53%(n=5),表明该方法精密度良好。
2.8 重复性试验
分别精密量取6份同一批号(20120718)CA4P囊泡400 μl,按供试品溶液的制备和测定法进行操作,测定CA4P含量,计算其RSD=0.85%(n=6),表明该方法重复性较好。
2.9 加样回收率试验

图2 检测限和定量限试验高效液相色谱图A.检测限试验;B.定量限试验Fig 2 HPLC chromatograms of detection limit and quantification limit testsA.detection limit test;B.quantification limit test
分别精密量取200 μl同一批号(20120718)已知CA4P含量的CA4P囊泡9份,置于5 ml棕色量瓶中,分3组,分别加入高、中、低相应量的CA4P对照品贮备液,用1 ml乙腈破坏囊泡使药物从囊泡中释放出来,用流动相定容至刻度,摇匀,用0.22 μm的微孔滤膜过滤,取续滤液,分别测定CA4P含量,计算回收率。结果平均回收率为96.72%,RSD=1.61%(n=3),表明该方法的准确度较好,详见表1。
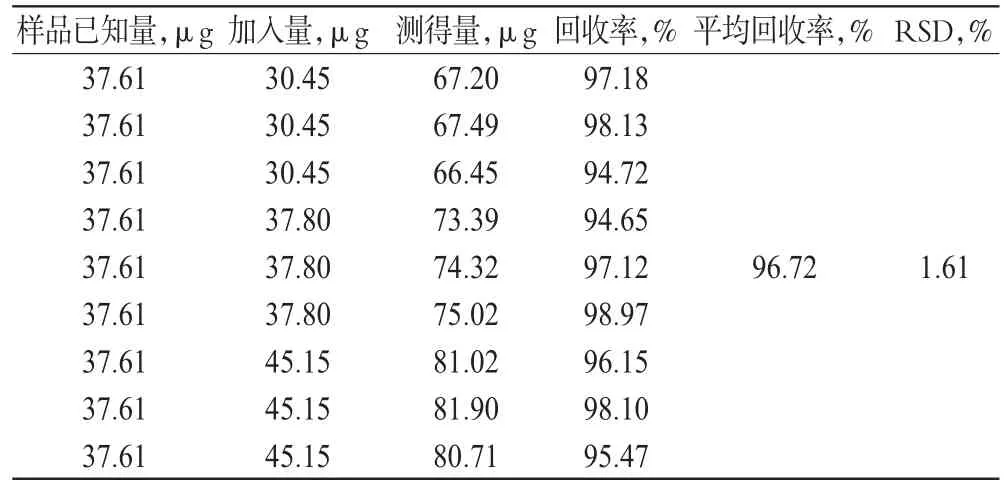
表1 回收率试验结果(n=3)Tab 1 Results of recovery test(n=3)
2.10 稳定性试验
取供试品溶液分别于配制后0~8 h内,每2 h测定1次,结果含量的RSD=1.75%(n=5),表明供试品溶液在配制后8 h内稳定。
2.11 样品含量测定
分别精密量取3批CA4P囊泡各3份,按供试品溶液处理方法制备供试品溶液,分别测定CA4P含量,结果见表2。
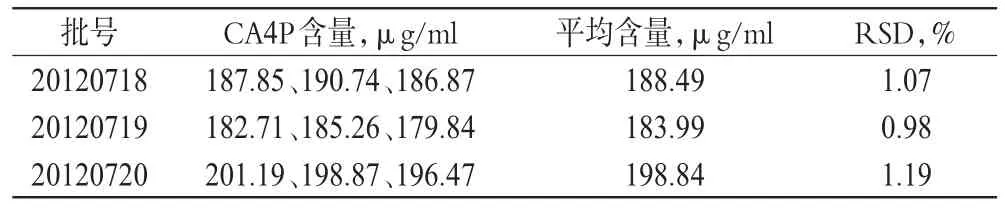
表2 3批样品中CA4P含量测定结果(n=3)Tab 2 Content determination results of CA4P in 3 batches of samples(n=3)
3 讨论
经扫描,CA4P在288 nm波长左右有最大吸收,与文献[5]基本一致,故选择288 nm为测定波长。与文献中流动相条件为磷酸二氢钾缓冲盐相比[10],本研究使用乙酸铵缓冲盐能防止峰拖尾、改善峰形,且更有利于保护色谱柱。因CA4P对光和热不稳定[6],在囊泡制备及含量测定过程中应注意避光操作。由于CA4P被包封于生物可降解高分子聚合物囊泡中,要测定包载在聚合物囊泡内的CA4P的量,必须将囊泡破坏使药物从囊泡中完全释放出来。因制备囊泡所用的高分子聚合物材料能溶于乙腈中,故采用乙腈破坏囊泡。经重复性试验及加样回收率试验证明,CA4P能够完全从囊泡中释放出来,方法的准确度及重复性均较好。
以上结果表明,RP-HPLC法测定聚合物囊泡中CA4P的含量,方法简便、准确度、精密度、重复性、稳定性好,可有效控制聚合物囊泡中CA4P的含量。
[1]Dark GG,Hill SA,Prise VE,et al.Combretastatin A-4,an agent that displays potent and selective toxicity toward tumor vasculature[J].Cancer Research,1997,57(10):1829.
[2]Prise VE,Honess DJ,Stratford MR,et al.The vascular response of tumor and normal tissues in the rat to the vascular targeting agent,combretastatin A-4-phosphate,at clinically relevant doses[J].Int J Oncol,2002,21(4):717.
[3]张亮,孟志云,孙文种,等.高效液相色谱法测定猕猴血浆中的风车子素A-4及其磷酸酯[J].药物分析杂志,2004,24(3):270.
[4]刘静,徐小平,张洁,等.绿原酸在兔体内对CA4P药动学的影响[J].中国药学杂志,2008,43(4):297.
[5]尹佳,张亚兰,莫毅,等.CA4P脂质体的制备工艺研究[J].华西药学杂志,2009,24(6):594.
[6]尹华熙,吴萍,徐小平,等.抗肿瘤新药CA4P及其有关物质的IR结构解析[J].华西药学杂志,2008,23(5):523.
