慢性低氧大鼠TRPC1表达与肺动脉收缩变化时间曲线关系
2014-05-15穆云萍焦海霞朱壮丽黄秋虹王瑞幸林默君
穆云萍,焦海霞,朱壮丽,戴 耄,黄秋虹,王瑞幸,林默君
(福建医科大学基础医学院生理学与病理生理学系,心血管科学研究室,福建福州 350108)
肺高压(pulmonary hypertension,PH)是指由异源性疾病和不同发病机制引起的以肺血管阻力进行性增加为主要特点的临床病理生理综合征[1]。典型的病理改变是血管张力增加、血管重塑和原位血栓形成,最终演化为危及生命的右心衰,是严重的慢性肺循环疾病[2]。现已证实,在慢性低氧(chronic hypoxia,CH)致 PH大鼠,其肺动脉平滑肌细胞(pulmonary artery smooth muscle cells,PASMCs)内升高的Ca2+浓度介导肺血管阻力进行性增加与PASMCs增生,是低氧性肺高压(hypoxic pulmonary hypertension,HPH)致病的关键因素[3]。而PASMCs上钙池操纵性 Ca2+通道(store-operated calcium channels,SOCC)与细胞内Ca2+稳态的维持密切相关[4]。经典瞬时感受器电位(canonical transient receptor potential,TRPC)家族编码的通道蛋白是SOCC的结构基础[5],SOCC可被 Ca2+池耗竭剂环匹阿尼酸(cyclopiazonic acid,CPA)激活,引起Ca2+池操纵性 Ca2+内流(store-operated calcium entry,SOCE)[3,6],参与平滑肌细胞生长或血管重塑等病理改变。前期工作表明,HPH大鼠模型 PASMCs上,TRPC1表达上调,它所介导的SOCE和血管收缩张力都明显增强[7],提示,由于上调的 TRPC导致SOCE所介导的肺动脉(pulmonary arteris,PAs)收缩增强,可能是HPH发生的重要原因。因此本研究利用 HPH大鼠模型,分别检测低氧 1、3、5、7、14、21 d等不同时间点,TRPC1表达与SOCE介导PAs收缩变化,通过比较两者时间上的相互关系,进一步阐明TRPC1/SOCE的上调,在PH发病过程中的作用。
1 材料与方法
1.1 CH肺高压模型的建立 清洁级♂ SD大鼠(200~220克),购自福建医科大学实验动物中心[SCXK(闽)2012-0001],随机分为慢性低氧0d组(NOR组)、1、3、5、7、14、21 d组,各组大鼠同时放于密封的有机玻璃饲养箱内,饲养箱内氧分压调至9.5% ~10.5%。
1.2 右心室内压测定 各组大鼠,用20%乌拉坦(0.5 g·kg-1)腹腔麻醉并进行肝素化处理,迅速经右颈外静脉行右心室插管术,记录大鼠平均右心室收缩压(mRVSP)。
1.3 右心室重量指数测定 右心室插管操作完成后,迅速取出大鼠的心、肺组织,用眼科剪沿房室交界剪去全部的左右心房及大血管,仅保留两侧心室,沿室间隔分离出右心室(RV),滤纸吸干后用分析天平称重,同法称取左心室与室间隔(LV+S)重量,计算 RVMI=RV/(LV+S)。
1.4 半定量RT-PCR检测TRPC1的 m RNA表达水平 测完右心室内压后,迅速取出肺叶,体视显微镜下分离出肺小动脉,纵向剪开,棉签去除内皮。在液氮中将标本充分研磨后,加 TRIzol试剂提取RNA,用紫外分光光度法测定RNA的浓度及纯度。按试剂盒的说明书进行RT-PCR。扩增条件:94℃2 min,94℃ 30 s,56℃ 30 s,68℃ 45 s,30个循环。引物序列:TRPC1上游引物5′-ATGGGACAGATGTTACAAGATTTTGGG-3′;下游引物 5′-AGCA-AACTTCCATTCTTTATCCTCATG-3′,扩增片段长度为402 bp。β-actin上游引物 5′-GGTGTGATGGTGGGTATGGGT-3′;下游引物 5′-CTGGG-TCATCTT-TTCACGGT-3′,扩增片段长度为240 bp。利用Genetool软件计算目的基因和内参(β-actin)条带的平均灰度值,以目的条带和β-actin的比值衡量各组目的基因表达的相对量。
1.5 肺动脉血管张力检测 测完右心室内压,取出肺叶,体视显微镜下分离出肺小动脉,剪成约3~5 mm的肺动脉环,去内皮后置于四腔浴槽中,给以1 g负荷,在RM6240多道生理记录仪(成都仪器厂)上描记张力曲线,平衡2 h。实验前先给予60 mmol·L-1KCl收缩血管,判断血管环是否具有活性。然后,加苯肾上腺素(phenylephedrine,PHEN,终浓度0.1μmol·L-1)预收缩达最大反应,再加乙酰胆碱(acetylcholine,ACh,10μmol·L-1),观察血管的舒张效应,以此来验证血管环去内皮情况[8]。血管环活性及去内皮度验证完毕后,观察环匹阿尼酸(cyclopiazonic acid,CPA,Sigma公司)对不同低氧时间点PAs的收缩效应,CPA诱发PAs的收缩作用以60 mmol·L-1KCl收缩效应的百分数来标准化。
1.6 统计学分析 数据以¯x±s表示,组间差异比较用one-way ANOVA方差分析,用Sigma Plot 11.0软件绘制曲线。
2 结果
2.1 慢性低氧上调mRVSP和RVM I CH诱发PH大鼠模型的过程中,分别在 0、1、3、5、7、14、21 d七个时间点检测大鼠的mRVSP和RVMI,并对其变化做时间依赖性曲线(Fig 1)。如图所示,RVSP在慢性低氧1 d就明显升高,7 d达到峰值(9.8±0.37)kPa,14 d后略有下降,直至21 d维持在 NOR组(4.0±0.16)kPa的两倍水平左右(9.7±0.66)kPa。但是,体循环动脉压和心率均没有差别[9],表明慢性低氧仅能够使右心室内压/肺动脉压增高。RVMI低氧3 d开始增加,一直维持增加状态,并未出现平台期,表明慢性低氧3 d后就开始发生右心重构,右心室肥大。的收缩效应约为NOR组的1.5倍(P<0.01),低氧5 d时收缩效应达最大值(90.2% ±15.1%,P<0.01),随后继续维持在较高水平,直至低氧21 d收缩效应仍较NOR组有明显增强,约为NOR组的2倍(87.2% ±7.1%,P<0.01),低氧增强 PAs血管环收缩效应的变化与TRPC1 mRNA表达量变化的时间曲线趋于一致,提示在低氧初期 TRPC1与SOCC介导的PAs的收缩效应即明显增强。
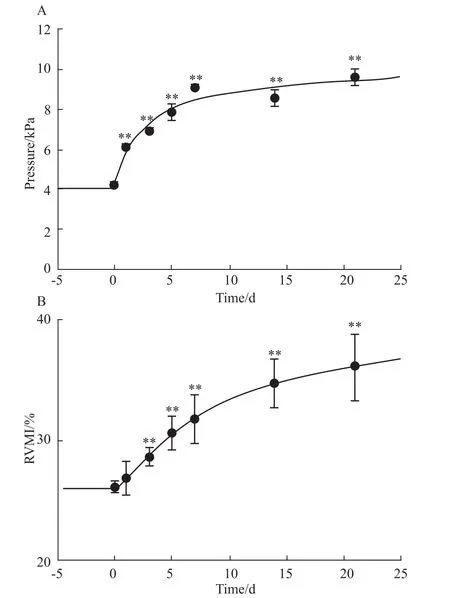
Fig 1 CH up-regulatesm RVSP and RVM IAverage values ofmRVSP at various time points after CH-induced(A,n=10 for each time point);average values of RVMIat various time points after CH-induced(B,n=10 for each time point)**P<0.01 vs control.

Fig 2 Time curve of expression of TRPC1 in PAsThe timetable ofmRNA expression of TRPC1(A).Semi-quantitative RT-PCR analysis of TRPC1 mRNA expression in PAs usingβ-actin as internal standard(n=3,**P<0.01).The time-dependent curve of TRPC1′s expression on CH rats(B)
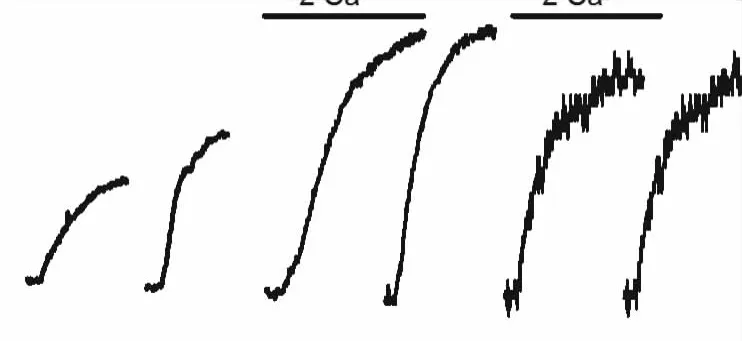
Fig 3 Time curve of vascular tone of PAsmediated by SOCETime course of change in CPA-induced PA contraction in CH-treated rats(A).The time-dependent curve of CPA(10μmol·L-1)response on CH rats(B),**P<0.01 vs CON.
3 讨论
临床PH的发生常继发于慢性阻塞性肺病等呼吸系统疾患,低氧是形成PH的重要因素,所以阐明HPH的发病机制一直是PH的研究热点。在低氧环境下,血管对活性物质的反应可明显增强,呈现慢性痉挛和过度肌化状态,导致低氧性肺血管收缩(hypoxic pulmonary vasoconstriction,HPV)。一定水平的HPV是机体为适应氧分压的降低而采取的一种保护措施,能够维持低氧肺泡区内适当的通气血流比值,起到通气代偿作用。但持续的HPV会引起肺循环外周阻力增加,最终形成HPH[10]。
本研究发现,RVMI在低氧3 d开始增加,随时间延长呈增高趋势,并且在低氧21 d内没有出现平台期;而RVSP在低氧1 d就明显升高,低氧7 d即达到峰值,并且在低氧21 d时维持在NOR组的两倍水平。以上结果提示持续低氧3 d即发生肺血管病理性改变,低氧7 d由于HPV加剧,导致肺血管重构,形成HPH,并随低氧时间延长建立稳定的PH模型。
先前的研究表明在慢性HPH大鼠,PASMCs上TRPC1表达与SOCE功能明显增高,升高的SOCE使PAs的收缩紧张度明显增强[7]。为了探讨TRPC1与SOCE功能的增强是否参与了HPH的发生和发展过程,本研究进一步检测了低氧不同时间TRPC1表达和PAs收缩变化的时间曲线。结果显示TRPC1 mRNA表达在低氧1 d即明显增加,低氧3 d时达最高水平,低氧21 d保持为NOR组的2倍。而低氧3 d时PAs的收缩效应开始较NOR组有所增强,至低氧5 d时收缩效应达到最大值,随后维持在较高水平,至低氧21 d时收缩效应也为NOR组的2倍。从结果中可以看到,低氧增强的PAs血管环收缩效应与TRPC1 mRNA表达变化,在时间上稍有延迟却又保持相同趋势。该结果提示TRPC1表达增强导致SOCE上扬,而后者又是介导PAs收缩增强的首要因素,最终引起HPH发生。
TRPC通道作为一类新发现的Ca2+通道,自其发现以来,就被认为与SOCE密切相关。研究表明,TRPC参与构成功能性SOCC,其介导产生SOCE调节细胞质与细胞器之间的Ca2+水平,TRPC过表达或表达下调直接影响SOCE水平[11],这与本研究观察到的两者在低氧不同时间点变化的同步性一致。在众多的TRPC通道中,TRPC1主要的功能为参与受体介导的Ca2+依赖的分泌和收缩过程。TRPC1是PASMCs以及体循环细胞中SOCE的主要成分[12],过表达大鼠PAs中的TRPC1会增强SOCE介导的血管收缩[13]。在增生的人PASMCs上,TRPC1表达与SOCE功能增强,经TRPC1反义寡核甘酸处理后,SOCE受到抑制。siRNA敲低大鼠PASMCs中的TRPC1会使thapsigargin激活的SOCE减弱,这些研究都提示TRPC1直接介导SOCE[3]。而本研究的实验结果也显示,在发生HPH的过程中,PASMCs上TRPC1 mRNA表达升高在前,随即由SOCE介导的PAs收缩增强,而持续的肺血管收缩引起RVSP升高,到CH的21 d时,形成了稳定的PH模型。
综上所述,本工作通过记录HPH模型建立的21 d内 RVSP、RVMI、TRPC1 mRNA以及 PAs收缩张力变化的时间曲线,明确了TRPC1表达上调与SOCE功能增强之间的关系,其在PH发生发展过程中的关键作用。
参考文献:
[1] Simonneau G,Robbins IM,Beghetti M,et al.Updated clinical classification of pulmonary hypertension[J].J Am Coll Cardiol,2009,54(1):S43-54.
[2] Hoette S,Jardim C,Souza R.Diagnosis and treatment of pulmonary hypertension:an update[J].J Bras Pneumol,2010,36(6):795-811.
[3] Lin M J,Leung G P,Zhang W M,et al.Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+channels in pulmonary arterial smoothmuscle cells:a novelmechanism of hypoxic pulmonary hypertension[J].Circ Res,2004,95(5):496-505.
[4] Sundivakkam P C,Freichel M,Singh V,et al.The Ca(2+)sensor stromal interaction molecule 1(STIM1)is necessary and sufficient for the store-operated Ca(2+)entry function of transient receptor potential canonical(TRPC)1 and 4 channels in endothelial cells[J].Mol Pharmacol,2012,81(4):510-26.
[5] Abramowitz J,Birnbaumer L.Physiology and pathophysiology of canonical transient receptor potential channels[J].FASEB J,2009,23(2):297-328.
[6] 承 伟,李 智.肺动脉高压模型大鼠肺动脉平滑肌细胞内钙的改变[J].中国药理学通报,2010,26(5):593-6.
[6] ChengW,Li Z.Alteration of[Ca2+]iin pulmonary artery smooth muscle of pulmonary artery hypertension rats[J].Chin Pharmacol Bull,2010,26(5):593-6.
[7] Liu X R,Zhang M F,Yang N,etal.Enhanced store-operated Ca(2+)entry and TRPC channel expression in pulmonary arteries ofmonocrotaline-induced pulmonary hypertensive rats[J].Am J Physiol Cell Physiol,2012,302(1):C77-87.
[8] 胡 莹,焦海霞,王瑞幸,郭立成.三七皂苷R1对肺高压大鼠模型肺动脉的舒张作用[J].中国药理学通报,2013,29(11):1572-6.
[8] Hu Y,Jiao H X,Wang R X,Guo L C.Vasodilation of notoginsenoside R1 on pulmonary arteries of pulmonary hypertensive rats[J].Chin Pharmacol Bull,2013,29(11):1572-6.
[9] Liu X R,Liu Q,Chen G Y,et al.Down-regulation of TRPM8 in pulmonary arteries of pulmonary hypertensive rats[J].Cell Physiol Biochem,2013,31(6):892-904.
[10]Kuhr FK,Smith K A,Song M Y,etal.Newmechanisms of pulmonary arterial hypertension:role of Ca(2+)signaling[J].Am JPhysiol Heart Circ Physiol,2012,302(8):H1546-62.
[11]El Boustany C,Bidaux G,Enfissi A,et al.Capacitative calcium entry and transient receptor potential canonical6 expression control human hepatoma cell proliferation[J].Hepatology,2008,47(6):2068-77.
[12]Yang X R,Lin M J,Sham JS.Physiological functions of transient receptor potential channels in pulmonary arterial smooth muscle cells[J].Adv Exp Med Biol,2010,(661):109-22.
[13]Kunichika N,Yu Y,Remillard CV,etal.Overexpression of TRPC1 enhances pulmonary vasoconstriction induced by capacitative Ca2+entry[J].Am J Physiol Lung Cell Mol Physiol,2004,287(5):L962-9.
