启动子工程改造大肠杆菌K4生产果糖软骨素
2014-05-04吴秋林杨爱华刘立明
吴秋林,刘 佳,杨爱华,刘立明
(1.江南大学 食品科学与技术国家重点实验室,无锡 214122;2.江南大学 工业生物技术教育部重点实验室,无锡 214122;3.江南大学 食品微生物制造工程实验室,无锡 214122)
硫酸软骨素(chondroitin sulfate,CS)作为糖胺聚糖类大分子酸性多糖,广泛存在于人体组织中,被誉为人体“软黄金”[1]。因其具有多种生理活性而作为膳食补充剂和保湿剂,被广泛应用于食品和化妆品领域[2-3]。硫酸软骨素二糖单体结构为4-GlcA-β-1,3-GalNAc-β-1(GlcA:葡 萄 糖 醛 酸,GalNAc:乙酰半乳糖胺),根据硫酸化发生位点的不同可分为 O、A、B、C、D、E 和 F 等类型,其中,动物来源的硫酸软骨素多为CS-A和CS-C[4]。目前,硫酸软骨素主要来源于动物提取[5]、酶法合成[6]和微生物发酵,但由于动物提取法存在生产周期长而酶法合成原料成本高等缺点,限制了其进一步应用。因此,微生物发酵法已成为最具经济效益和开发潜力的生产工艺[7]。据研究,硫酸软骨素能以荚膜多糖的形式存在于巴斯德杆菌(Pasteurella multocida)、大肠杆菌(Escherichia coli)和枯草芽胞杆菌(Bacillus subtilis)等 微 生 物 的 细 胞 壁 中[8-10]。 虽 然,P.multocida能合成软骨素类荚膜多糖,但是其为家禽霍乱致病菌。近年来,研究重点转向利用来源于P.type F的软骨素合成酶pmCS进行酶法转化生产软骨素多糖链,但由于高昂的单糖供体成本,目前尚无量产报道[9,11-12]。美国 Amano Enzyme 公司筛选获得1株可直接生产硫酸软骨素的纳豆枯草芽胞杆菌(B.subtilis natto),但其产量仅为 0.24 g/L[10]。大肠杆菌K4(E.coli K4)能合成软骨素类似物作为其荚膜多糖(capsular polysaccharide,K4CPS),即果糖软骨素(4-β-GlcA(Fru)-1,3-β-D-N-GalNAc-1)[8],经脱果糖和硫酸化修饰等步骤可获得硫酸软骨素[13]。此外,大肠杆菌具有遗传背景清楚、荚膜多糖合成与转运机制清晰等优点而成为研究硫酸软骨素发酵的主要菌株。Schiraldi等[14]采用微膜生物反应器高密度培养大肠杆菌K4,可使K4CPS产量高达4.73 g/L,但比细胞得率仅为0.13 g/g(以1 g干细胞质量中含K4CPS量计)。
目前,根据遗传学特征、生物合成方式及其分子结构等特性,可将大肠杆菌荚膜多糖分为4大类,即 group 1 ~group 4[15]。大肠杆菌 K4 属于 group 2,其K4CPS合成基因簇包括 region 1、region 2和region 3 3 部分(图1)[16]。其中 region 1 和 region 3主要参与新合成多糖链的修饰、转运与定位[17],而region 2包括kfoA~G等7个基因和1个插入序列IS2,主要编码多糖及其前体合成相关的酶[18]。此外,group 2荚膜多糖合成基因簇的转录由2个σ70启动子所调控,分别是位于region 1上游224 bp的PR1和region 3上游741 bp的PR3。PR1转录产生1条约8 kb的多顺反子,随后再转录出1条约1.3 kb的kpsS转录子[19]。而 PR3的转录可从region 3延伸至region 2,此过程需要RfaH蛋白与PR3启动子3'端非翻译区(untranslated region,UTR)713 bp处ops序列的结合,在转录过程中与RNA聚合酶复合物相互作用,调节region 2转录的正常进行。ops序列的缺失或rfaH基因的突变均可减弱荚膜多糖的合成[20]。同时,UTR具有减弱PR3活性的作用,整个741 bp UTR片段的缺失可显著提高PR3的转录活性[21]。因此,为了进一步提高K4CPS产量,笔者通过Red同源重组技术对UTR进行敲除,以增强PR3启动子的活性,从而促进K4CPS的合成。
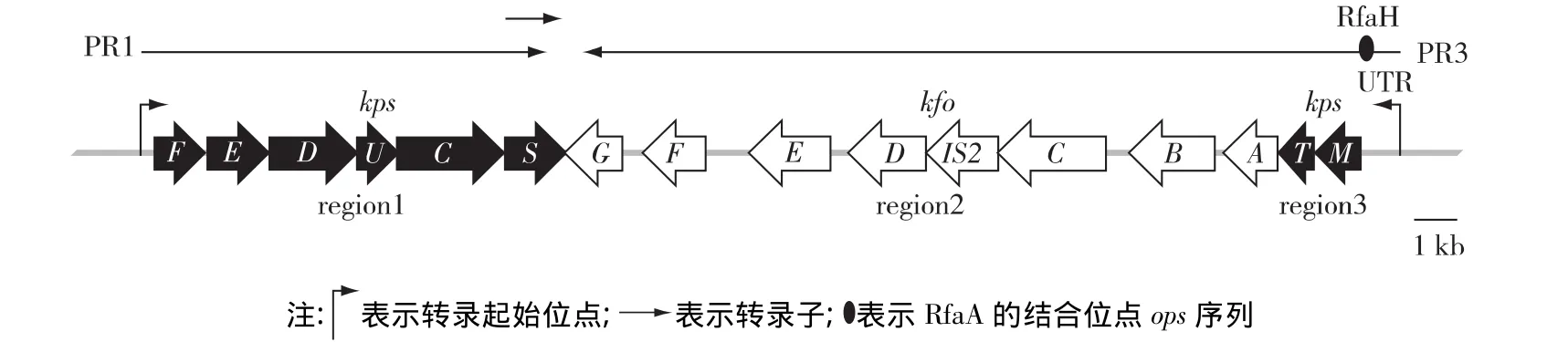
图1 大肠杆菌K4荚膜多糖合成基因簇的转录[21]Fig.1 Transcription of E.coli K4 CPS gene cluster[21]
1 材料与方法
1.1 菌株和质粒
大肠杆菌K4(E.coli O5:K4:H4)购买于美国典型培养物收藏中心(ATCC),保藏编号为ATCC 23502;大肠杆菌JM109为笔者所在实验室保藏;质粒pKD46(含有受ParaB启动子调控的exo、bet和gam基因)、pKD4(含有卡那霉素抗性)、pCP20(含有翻转酶重组酶基因)均由美国Purdue大学B.L.Wanner惠赠;克隆质粒pMD18-T购买于Takara公司。
1.2 培养基和培养条件
Red同源重组过程和种子培养基均用LB培养基。根据菌株和所用质粒的抗性在培养基中添加抗生素,氨苄青霉素50 ~100 mg/L,卡那霉素25~50 mg/L。摇瓶发酵培养基(g/L):甘油20,大豆蛋白胨 1,KH2PO42,K2HPO49.7,(NH4)2SO43,柠檬酸三钠0.5,MgCl20.1。培养条件为37℃、200 r/min培养24 h。
1.3 Red 同源重组[22]
1.3.1 同源重组引物设计与敲除框的构建
利用软件Primer Premier 5设计同源重组引物,其设计位点如图2。扩增pKD4卡那霉素抗性基因kan的引物P3(5'-ATGATGTGATCCTAATCTCTTCAGGTATGCTACCGCCCCTGGCTTAACTGTGTAGGCTGGAGCTGCTTCG-3')、P4(5'-TGTATTTTGTGTAGTCTGGAAATTAGTAAAATTCCTGGAGATAATCAGAAATGGGAATTAGCCATGGTCC-3')和P8(5'-ACTTTCTGGACTTCAAATCCACTTCTTGCCATTTGATGATGTGATCCTAGTGTAGGCTGGAGCTGCTTCG-3')分别由2部分组成,靠近5'端的序列与PR3待敲除序列两翼序列同源,靠近3'端斜体加粗的序列与kan基因两侧序列互补。在PR3启动子UTR两侧,设计引物P1(5'-ATCAGCGTCTCAAGCAGTG-3')、P2(5'-AGGAACTTCGAAGCAGCTCCAGCCTACACAGTTAAGCCAGGGGCGGTA-3')、P7(5'-ATTCTCTAGAAAGTATAGGAACTTCGAAGCAGCTCCAGCCTACACTAGGATCACATCATCAAATGGCAAG-3')用于扩增敲除框的右翼,P2和P7靠近5'端的序列与kan基因序列同源,靠近3'端斜体加粗序列与UTR+698 bp和+724 bp互补。P5(5'-ATATTCATATGGACCATGGCTAATTCCCATTTCTGATTATCTCCAGGAAT-3')和P6(5'-CTTAGCCCAATGGTGAGT-3')用于扩增敲除框的左翼,P5靠近5'端的序列与kan基因序列同源,靠近3'端斜体加粗序列与UTR+36 bp互补。所用引物由上海生工生物工程公司合成。
以大肠杆菌K4基因组和pKD4为模板,利用上述引物可扩增得到敲除框的左翼片段、右翼片段和kan基因片段。将此3种片段以等摩尔浓度混匀后作为模板,以P1和P6为引物进行融合PCR,最终得到带有300~400 bp同源臂的敲除框。
1.3.2 感受态的制备

图2 同源重组引物设计位点Fig.2 Localsites of primers used for Red recombination
将培养12 h含有 pKD46的大肠杆菌 K4,以1%(体积分数)的接种量转接至50 mL LB培养基中,30℃培养1 h后加入0.5 mL 1 mol/L的L-阿拉伯糖,培养至OD600为0.5~0.6。将50 mL培养液冰浴30 min后,4℃、8 000 r/min离心3 min。收集菌体,用预冷无菌水洗涤菌体1次,用预冷10%(体积分数)甘油洗涤菌体2次,最后加入200 μL预冷10%甘油重悬菌体制备成感受态细胞。
1.3.3 电击转化与筛选
取100~200 ng敲除框DNA与50 μL感受态细胞混匀,冰浴10 min。用 Bio-Rad公司 MicroPulser电穿孔仪进行电击转化,0.1 cm电击杯,转化参数为1.8 kV、5 ms、25 F、200 Ω。电击后加入 1 mL LB培养基,37℃预培养1 h,涂布于含有卡那霉素的LB平板。37℃倒置培养12~16 h,筛选阳性转化子。转化子在43℃培养12 h筛选氨苄青霉素缺失菌株。然后,按照1.3.2的方法制备感受态(不加L-阿拉伯糖),电击转化质粒pCP20。转化液涂布于含有氨苄青霉素的LB平板,37℃培养12 h,筛选阳性转化子。转化子再转入43℃培养12 h筛选氨苄青霉素和卡那霉素同时缺失的突变株。
1.4 荧光定量PCR
发酵培养突变株和原菌至对数生长末期,取约109个细胞,按照细菌总RNA提取试剂盒(天根)提取总RNA,并设3个重复。经非变性凝胶电泳检测后通过反转录试剂盒(Bio-Rad公司)得到cDNA。以cca和idnT基因作为双内参基因,对kpsE、kfoC和kpsM基因的表达水平进行相对定量,所用扩增引物由Beacon Designer 7设计(表1)。荧光定量PCR反应体系为20 μL(3 个平行):无菌水6 μL、cDNA 2 μL、引物各 1 μL、SYBR Green supermix(Bio-Rad 公司)10 μL。PCR反应程序:95℃预变性30 s;95℃变性5 s,50℃退火10 s,40个循环;融解曲线:65℃到95℃,每上升0.5℃保持10 s。反应结果利用Bio-Rad CFX Maneger软件采用 2-ΔΔCt法进行分析。
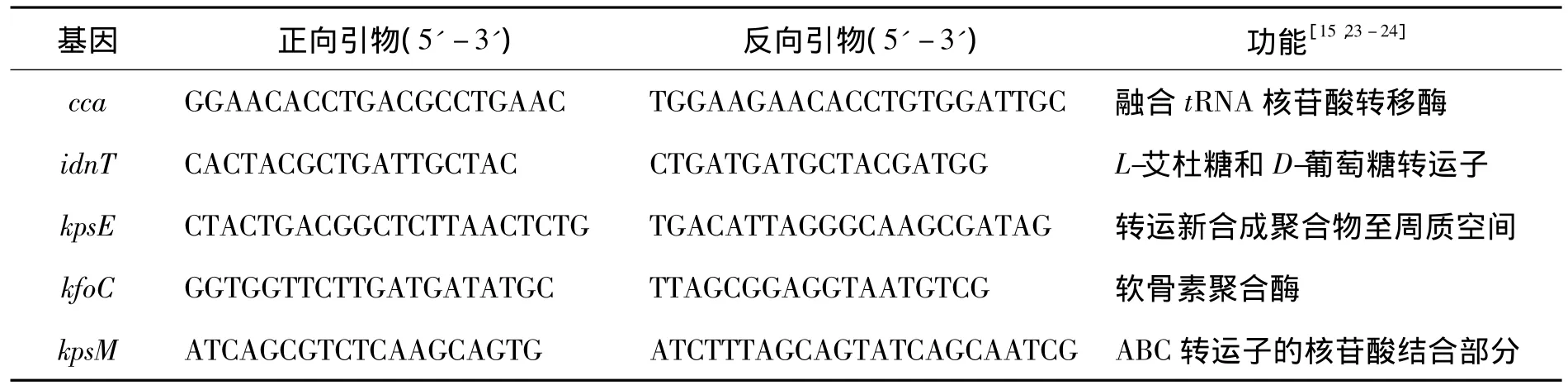
表1 荧光定量PCR所用引物Table 1 Primers used for quantitative real time PCR
1.5 分析方法
1.5.1 K4CPS含量的测定
发酵液8 000 r/min离心3 min收集上清液,取少量上清液经蒸发浓缩后,加入1 mL无水乙醇洗去杂质。离心所得沉淀加入适量去离子水重悬,用于K4CPS含量的测定。K4CPS含量用咔唑法测定[25]:取5 mL浓H2SO4(含有9.5 g/L四硼酸钠)与1 mL样品混匀,煮沸10 min;迅速冷却至室温,加入200 μL咔唑溶液(0.125 g咔唑溶于乙醇中)振荡混匀后煮沸15 min,迅速冷却至室温。在530 nm处测定吸光值。以硫酸软骨素A(Sigma公司)为标准品,质量浓度梯度为25~125 mg/L,最终所得K4CPS含量(mg/L)=标准曲线得到浓度×稀释倍数×1.12。
1.5.2 甘油浓度的测定
采用高效液相色谱仪UltiMate 3000(Dionex公司)检测上清液中甘油的含量,条件:分离柱Aminex HPX-87H column(Bio-Rad公司),柱温35℃,进样量 20 μL,流动相为体积分数 0.027 5%H2SO4溶液,流动相流速 0.6 mL/min,示差折光检测器(RID)。
1.5.3 细胞干质量的测定
将发酵液稀释至适当倍数,在600 nm处测定其吸光值,细胞干质量(DCW)=吸光值×稀释倍数 ×0.412。
2 结果与分析
2.1 大肠杆菌K4 PR3启动子的克隆和序列同源性分析
以大肠杆菌K4基因组为模板,以P1和P6为引物PCR扩增得到1条约1.4 kb的片段。纯化后连接至pMD18-T载体,转化E.coli JM109感受态细胞,涂布于含有氨苄青霉素的LB平板上,筛选得到带有PR3启动子的重组子。重组质粒pMD18-PR3经测序分析,插入片段长度为1 403 bp,包括PR3启动子及其3'端的741 bp UTR和kpsM基因的5'端。通过Blast比对分析发现:大肠杆菌 K4与K5的PR3启动子的同源性为96.54%,转录起始位点由A变为G。
2.2 PR3启动子3'端UTR缺失突变株的构建与验证
以P3、P4(或 P8)为引物,pKD4为模板,扩增kan基因,PCR产物约1.5 kb;以P1、P2(或P7)为引物,大肠杆菌K4基因组DNA为模板,扩增UTR左侧序列,约 400 bp;以 P5、P6为引物,K4基因组DNA为模板,扩增UTR右侧序列,约300 bp。三段PCR产物经过融合PCR分别得到2条约2.2 kb的敲除框。按照1.3 Red同源重组技术对UTR区进行缺失突变,得到4株UTR区加长或缩短的突变菌株。敲除流程如图3(a)所示。由图3可知:由2条敲除框经过第一次同源重组将kan基因整合到UTR区分别得到突变株PR301::kan和PR303::kan。随后在翻转酶的作用下,kan基因两侧的FRT位点发现第二次同源重组,kan基因丢失得到突变株PR301和PR303。表2为上述突变菌株的特性,其中菌株PR301::kan(PR301)在保留ops序列的同时延长(缩短)UTR序列;而PR303::kan(PR303)则是缺失ops序列的同时延长(缩短)UTR序列。引物P1和P6进行扩增,根据重组前后序列长度的变化进行验证(图3b),并将PCR产物送至华大基因测序,结果与预期相符。
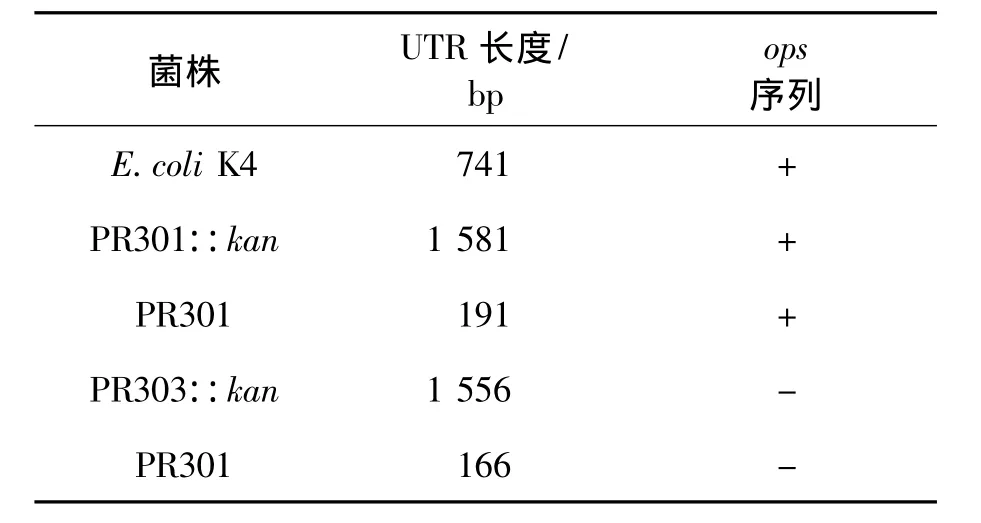
表2 突变株特性Table 2 Characteristics of mutants

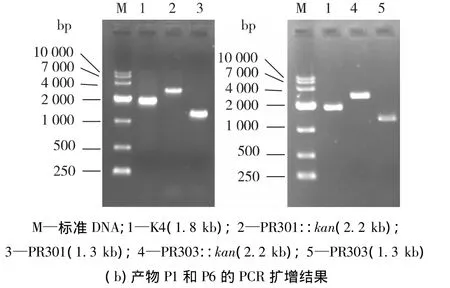
图3 UTR缺失突变株的构建及PCR鉴定Fig.3 Construction of mutants with UTR deletion and PCR analysis
2.3 UTR缺失突变对K4CPS合成的影响
为研究UTR缺失突变后对K4CPS生产的影响,将原菌E.coli K4与UTR突变株进行摇瓶发酵培养24 h,检测其细胞生长、甘油利用和K4CPS合成的情况,结果如表3所示。由表3可知:与原菌相比,4株突变株的生物量和甘油利用率变化并不显著。其中,突变株 PR301::kan和 PR301的K4CPS产量与E.coli K4也无明显的变化,这一结果表明,在ops序列存在的情况下,UTR序列的延长或缩短并不能影响K4CPS产量。然而,对于菌株PR303::kan,在ops序列缺失和延长UTR序列的双重调控下,可导致K4CPS产量的降低,比原菌下降了16%;反之,在ops序列缺失的基础上,缩短UTR序列至166 bp可显著提高菌株PR303生产K4CPS的能力,使其产量达751 mg/L,而比细胞得率高达0.214 g(以1 g干细胞质量中含K4CPS量计),比原菌分别提高了46% 和51%。上述结果表明:缺失 ops序列且缩短UTR能有效地提高K4CPS产量;ops序列或RfaH蛋白并不是K4CPS合成的必需蛋白。
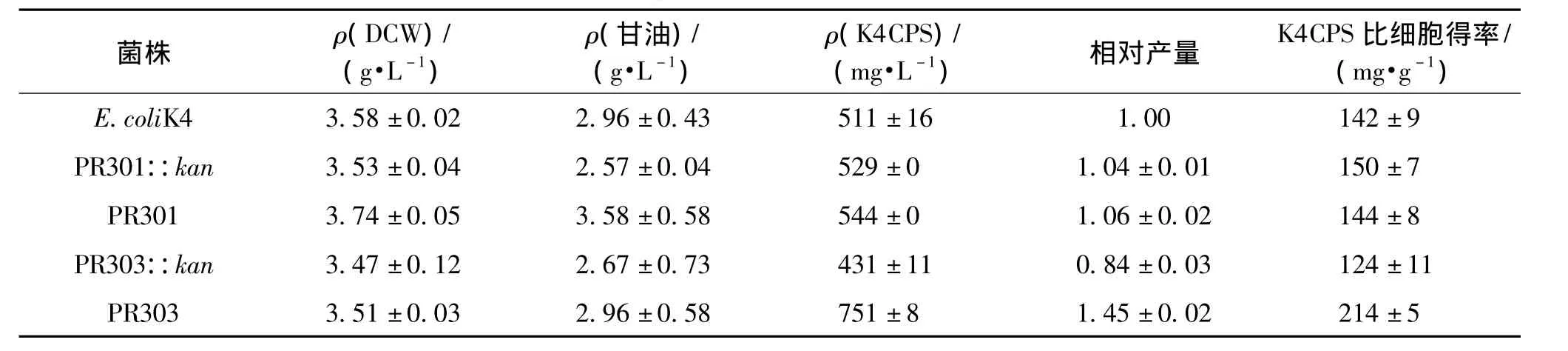
表3 突变株与原菌的K4CPS产量对比Table 3 K4CPS production in mutants and E.coli K4
2.4 UTR缺失突变对PR3启动子强度的影响
为了解析UTR突变如何影响K4CPS合成,选取K4CPS合成基因簇中region 1、region 2和region 3的代表性基因kpsE、kfoC和kpsM,以cca和idnT基因为双内参基因,通过荧光定量PCR对kpsM、kfoC、kpsE基因的表达水平进行相对定量,结果见图4。由图4可知:与原菌相比,在突变菌株PR301::kan和PR301中,3个基因的表达水平均无显著变化,而在PR303::kan中,kpsM和kfoC基因的表达水平则分别下降为原菌的58% 和68%;但在PR303中kpsM和kfoC基因的表达水平则分别提高了1.04和1.16倍。此外,4株突变菌的kpsE基因表达水平均无显著变化,说明UTR的突变对region 1基因的转录表达没有明显的影响。因此,从基因转录水平上研究表明,在ops序列存在的情况下,UTR序列的延长或缩短都难以影响PR3启动子的强度,而ops序列缺失后,UTR区的延长能减弱PR3启动子的强度,UTR区的缩短则能有效地加强PR3启动子的强度。上述结果表明,RfaH蛋白的与ops序列的结合会影响UTR区对PR3启动子的调控作用,但其具体的作用机制有待进一步研究。

图4 荚膜多糖合成基因簇基因表达水平分析比较结果Fig.4 Analysis of expression levels of genes in capsule gene cluster
3 讨论
目前,国内外研究人员已从多方面对发酵法生产硫酸软骨素开展了卓有成效的研究工作。然而,由于微生物自身代谢的经济型生存本能和工业生产环境与自然环境的巨大差异,使硫酸软骨素的产量、产率和生产强度均难以达到工业化生产的要求。因此,利用基因工程手段获得硫酸软骨素高产菌株已成为硫酸软骨素研究领域的热点和重点[7],如过量表达K4CPS合成途径中的软骨素酶合成基因可有效提高 K4CPS产量[26]。此外,过量表达UDP-葡萄糖脱氢酶能够使荚膜多糖合成前体UDPGlcA的产量提高3倍,但意外的是,K4CPS的产量并没有提高,反而显著地降低了3倍[27]。其原因可能是硫酸软骨素等荚膜多糖的合成涉及单糖合成、核糖供应、酶动力学反应以及膜转运蛋白等多个环节,仅对单一或者2~3个基因进行代谢工程改造难以显著提高K4CPS生产效率。因此,如要提高硫酸软骨素的产量,需要同时对多个基因的表达水平进行调控。
同时提高胞内多个基因表达水平的另一策略是过量表达全局转录调控蛋白,如增加fis基因(编码转录调控蛋白)拷贝数和突变σ因子均能提高透明质酸的产量[28-29]。但需考虑的是对具有潜在致病性的生产菌株进行全局转录蛋白调控时,可能会引起多种毒性因子(如脂多糖)的变化。在对荚膜多糖合成基因簇转录调控子进行详尽分析的基础上,采用Red同源重组技术对荚膜多糖合成基因簇PR3启动子进行改造,在ops序列缺失的基础上缩短PR3启动子3'端UTR的长度,增强PR3启动子的活性。使region 2和region 3的表达水平同时得到了提高,从而显著提高了K4CPS的生产效能,其比细胞得率达到214 mg/g,为目前报道的最高水平。分析其原因可能为:region 2中负责编码K4CPS多糖聚合酶(KfoC)及其单糖前体UDP-GalNAc与UDP-GlcA合成相关酶(分别为KfoA和KfoF)表达水平的提高,增强了胞质中单糖前体合成和多糖聚合的代谢强度;由于region 3中KpsM和KpsT蛋白组成的ABC-A2型转运系统的加强,加速了K4CPS多糖链从胞内向胞外的转运;同时提高胞内K4CPS合成强度与跨膜转运速率可能更有利于胞外K4CPS的高效积累。这一研究表明,通过启动子工程从转录水平对荚膜多糖合成基因簇进行整体调控,能显著促进荚膜多糖的合成,为相关研究提供了新的代谢工程策略。
[1] 翟中和,王喜忠,丁明孝.细胞生物学[M].3版.北京:高等教育出版社,2008.
[2] Zhou S.Health food useful for improving water content of skin,obtained bymixingcomponentsincludinghyaluronicacid,chondroitin sulfate,fish collagen,vitamin C powder,glycine,proline,extract of grape seeds and starch:CN,102210425A[P].2011-10-12.
[3] WadaT,Mano T,TanouchiM.Composition usefulin pharmaceuticals and food-drinks for treating asthenopia,comprises mixture of chondroitin sulfate:WO,2011136159A1[P].2011-03-11.
[4] Schiraldi C,Cimini D,DeRosa M.Production of chondroitin sulfate and chondroitin[J].Appl Microbiol Biotechnol,2010,87(4):1209-1220.
[5] Lauder R.Chondroitin sulphate:acomplexmoleculewith potential impacts on a wide range of biological systems[J].Complement Ther Med,2009,17(1):56-62.
[6] DeAngelis P.Chondroitin synthase gene and methods of making and using same:US,7569386B2[P].2005-06-24.
[7] 吴秋林,刘立明,陈坚.发酵法生产硫酸软骨素的研究进展[J].生物工程学报,2012,28(11):1281-1293.
[8] Rrodriguez M L,Jann B,Jann K.Structure and serological characteristics of the capsular K4 antigen of Escherichia coli O5:K4:H4,a fructose-containing polysaccharide with a chondroitin backbone[J].Eur J Biochem,1988,177(1):117-124.
[9] DeAngelis P L,Gunay N S,Toida T,et al.Identification of the capsular polysaccharides of type D and F Pasteurella multocida as unmodified heparin and chondroitin,respectively[J].Carbohydr Res,2002,337(17):1547-1552.
[10] Jolly J,Klimaszewski K,Nakanishi Y,et al.Microbial-derived chondroitin sulfate:US,20100063001A1[P].2009-06-03.
[11] Chong B,Blank L,Mclaughlin R,et al.Microbial hyaluronic acid production[J].Appl Microbiol Biotechnol,2005,66(4):341-351.
[12] Vann W,Schmidt M,Jann B,et al.The structure of the capsular polysaccharide(K5 antigen)of urinary-tract-infective Escherichia coli 010:K5:H4:a polymer similar to desulfo-heparin[J].Eur J Biochem,1981,116(2):359-364.
[13] Bedini E,DeCastro C,DeRosa M,et al.A microbiologicalchemical strategy to produce chondroitin sulfate A,C[J].Angew Chem Int Ed,2011,50(27):6160-6163.
[14] Schiraldi C,Restaino O,Cimini D,et al.High cell density cultivation of Escherichia coli K4 in a microfiltration bioreactor:a step towards improvement of chondroitin precursor production[J].Microb Cell Fact,2011,10(1):10-19.
[15] Whitfield C,Roberts I.Structure,assembly and regulation of expression of capsules in Escherichia coli[J].Mol Microbiol,1999,31(5):1307-1319.
[16] Whitfield C. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli[J].Annu Rev Biochem,2006,75:39-68.
[17] Roberts I,Mountford R,Hodge R,et al.Common organization of gene clusters for production of different capsular polysaccharides(K antigens)in Escherichia coli[J].J Bacteriol,1988,170(3):1305-1310.
[18] Ninomiya T,Sugiura N,Tawada A,et al.Molecular cloning and characterization of chondroitin polymerase from Escherichia coli strain K4[J].J Biol Chem,2002,277(24):21567-21575.
[19] Simpson D A,Hammarton T C,Roberts I S.Transcriptional organization and regulation of expression of region 1 of the Escherichia coli K5 capsule gene cluster[J].J Bacteriol,1996,178(22):6466-6474.
[20] Stevens M P,Clarke B R,Roberts I S.Regulation of the Escherichia coliK5 capsule gene clusterby transcription antitermination[J].Mol Microbiol,1997,24(5):1001-1012.
[21] Xue P,Corbett D,Goldrick M,et al.Regulation of expression of the region 3 promoter of the Escherichia coli K5 capsule gene cluster involves H-NS,SlyA,and a large 5'untranslated region[J].J Bacteriol,2009,191(6):1838-1846.
[22] Datsenko K A,Wanner B L.One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products[J].PNAS,2000,97(12):6640.
[23] Lidholt K,Fjelstad M.Biosynthesis of the Escherichia coli K4 capsule polysaccharide:a parallelsystem for studies of glycosyltransferases in chondroitin formation[J].J Biol Chem,1997,272(5):2682-2687.
[24] Zhou K,Zhou L,Lim Q,et al.Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR[J].BMC Mol Biol,2011,12(1):18.
[25] Zanfardino A,Restaino O,Notomista E,et al.Isolation of an Escherichia coliK4 kfoC mutantover-producing capsular chondroitin[J].Microb Cell Fact,2010,9(1):34-41.
[26] Cimini D,DeRosa M,Viggiani A,et al.Improved fructosylated chondroitin production by kfoC overexpression in E.coli K4[J].J Biotechnol,2010,150(3):324-331.
[27] Roman E,Roberts I S,Lidholt K,et al.Overexpression of UDP-glucose dehydrogenase in Escherichia coli results in decreased biosynthesis of K5 polysaccharide[J].Biochem J,2003,374(3):767-772.
[28] Yu H,Tyo K,Alper H,et al.A high-throughput screen for hyaluronic acid accumulation in recombinant Escherichia coli transformed bylibraries ofengineered sigma factors[J].Biotechnol Bioeng,2008,101(4):788-796.
[29] Steen J A,Steen J A,Harrison P,et al.Fis is essential for capsule production in Pasteurella multocida and regulates expression of other important virulence factors[J].PloS Pathogens,2010,6(2):e1000750.
