高通量测序序列比对研究综述
2014-04-29高静焦雅张文广
高 静 焦 雅 张文广

摘要:高通量测序技术的飞速发展,给生物信息学带来了新的机遇和挑战,第二代测序序列数量多、长度短使得原来的序列分析手段不再适用。近几年来,针对高通量测序的序列分析算法和软件日益增多,目前已有上百种,导致选择合适的软件成为一个难题。对第二代测序的测序类型、序列类型以及分析算法进行了总结和归纳,对现今常用的分析软件的序列的类型、长度以及软件应用算法、输入/输出格式、特点和功能等方面做了详细分析和比较并给出建议。分析了现今测序技术和序列分析存在的问题,预测了今后的发展方向。
关键词:高通量测序:序列比对;序列作图;序列比对工具
中图分类号:Q-31
文献标识码:A
文章编号:1007-7847(2014)05-0458-07
以Roche/454焦磷酸测序(2005年)、lllumina/Solexa聚合酶合成测序(2006年)和ABI/SOLiD连接酶测序(2007年)技术为代表的第二代测序技术与Sanger测序相比,共有的突出特征是单次运行产出序列数据量大,故而又被通称为高通量测序技术。高通量测序技术大大降低了测序的时问和成本,因而得到了广泛应用。但随之而来的短序列(short read)为基因数据分析带来了新挑战。目前对短序列一个常用的分析方法则是将已有基因组序列作为参考基因序列(reference),将短序列与参考基因序列进行序列比对,并在参考基因序列上进行定位,这个过程称为mapping。序列比对是基冈数据分析最基本的手段,对其进行研究具有重要意义。
1 高通量测序概述
1.1 测序分类
大规模、低成本、快速的高通量测序技术被广泛应用于生物研究的各个方面,当前主流的测序分为:1)基因组学的全基因组de novo测序、全基因组重测序、外显子&目标区域测序、简化基因组测序;2)转录组学的转录组测序、数字基因表达谱、小RNAs测序、降解组测序和长链非编码BNA洲序;3)表观基囚组学的全基因组Bisulfile甲基化测序、RRBS、MeDIP测序等。
1.2 测序数据类型
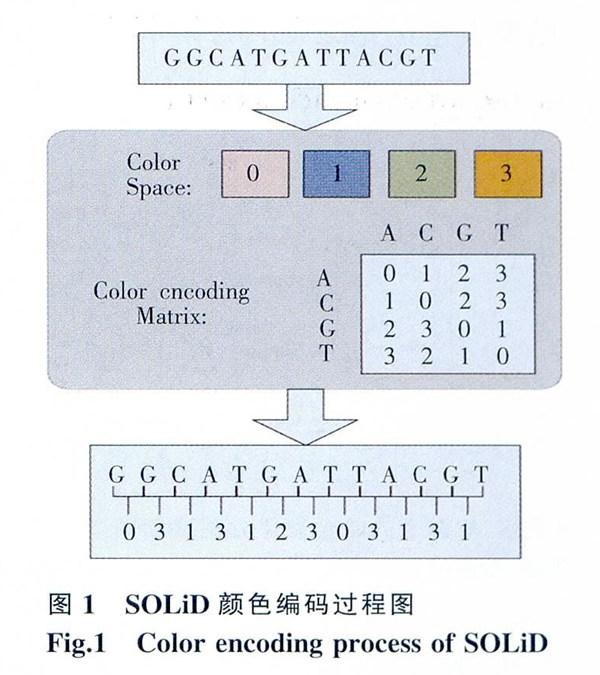
现在比较常见的测序方式有:single -read、paired-end、Mate-pair、color-space。1)Single-read即为单末端测序,在测序前先将DNA样本进行片段化处理形成200~500 hp的片段,且将引物序列连接到DNA片段的一端,然后末端加上接头。这种方法较简单,但是它只有一端有拼接信息,不利于拼装;2)Paired-end称为双末端测序或配对末端测序,它是指在构建待测DNA文库时,在基因片段的两端都加上测序引物结合位点再进行测序。双末端测序是在片段两端都加上接头,在序列拼装的时候更容易定位片段;3)Male-pair也属于双末端测序,与paired-end不同的是它将基因随机打成特定为2~10 kb的片段,然后经末端修复,生物素标记和环化等实验步骤后.再把环化后的片段打断成400~600 bp的片段并标记测序。因为经过了环化步骤,Mate-pair比paired-end方式测得的序列片段更长,从而可以将基因序列中大量的repeat包含在内,减少拼接难度;4)Color-space read是ABI公司的SOLiD测序仪检测出的read,SOLiD可以同时检测两个相邻的碱基,并利用颜色空间的4种不同的颜色对两个碱基编码。对于这种编码方式,只要知道颜色编码对应的序列上任何一个位置的碱基类型,就可以将颜色编码解码为原来的碱基序列,图l为颜色编码过程。
2 序列比对方法
对于百万条甚至上亿条短序列在reference上的定位,传统的动态规划算法不能满足我们的要求。为了加快序列比对的速度,应用启发式算法是必然趋势。同前,绝大多数的启发式算法都是通过建立索引加快比对速度。建立索引就是用一个辅助的数据结构存储待比对序列,这种数据结构能够将序列中符合一定规则的子序列凸显出来。常用的数据结构有哈希表和后缀树两种。
2.1 基于种子的哈希表索引方法
这种方法多用于数据库搜索或read在参考序列上的mapplng,以哈希表形式建立序列的索引,这类算法的代表软件是SSAHA,基于种子的哈希表方法具体步骤:
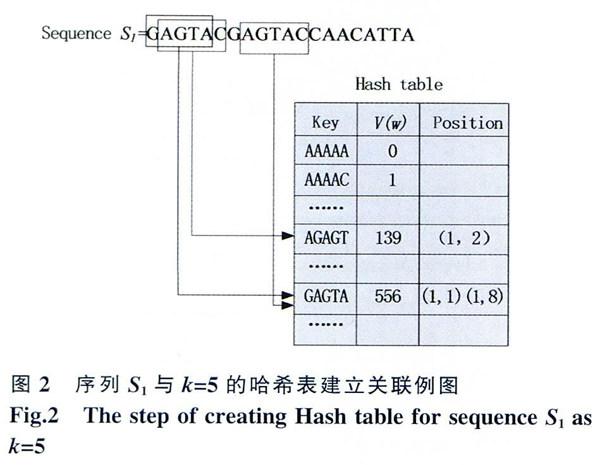
第一步,建立哈希表。
DNA序列由A、C、T、G四种脱氧核苷酸组成,长度为k的连续片段也就是“种子”有4K种可能将4K种子存放在哈希表中,我们用一个公式将所有种子唯一标识,将4个碱基的值f(x)转换为二进制数据表示,只占用2 Bit的内存。
第二步,将数据库中的序列与哈希表关联
设一个含有n条DNA序列的数据库D={S1,S2,…,Sn},将数据库中的每条序列分解连续种子w,其长度k=5,从头开始每次偏移一位直到序列结束,那么长度为l的序列有(l-k+1)个种子。并将每一个“种子”所在的序列,N(N=l,2,…,n)和在序列中的顺序号L (L=1,2,…,l)在对应的哈希表中的“种子”以二维数组方式记录下来(N,L)。将S1
第三步,查询序列Q也分解成长度为k的种子并计算其标识号,在哈希表中找到数据库中与之匹配的所有位置。
为了让算法更高效,算法在各个方面进行了改进:1)“种子”长度,允许错配和indel的数量等因素直接影响比对结果的准确性、敏感度、时间和空间花费等指标,所以研究者们一直致力于对“种子”形式和选择的改进中。Blastn、Blat、SOAP、SeqMap、MAQ、SHRiMP使用不同的种子模型,如空位种子、空位种子组(同时使用多个种子模型)等;2)-些软件如MAQ、RMAP、SeqMap、ZOOM等将reads序列建立索引,而一些软件将参考序列数据库建立索引;3)设置阈值F将哈希表中出现频率低于F的“种子”删去。
2.2 基于前/后缀树的索引方法
后缀树是一种重要的数据结构,AVID、MUMmer、MUMmer等序列比对算法都是基于后缀树的这个结构,但与两序列相似性比对时用于寻找最大公共子序列不同,read在参考序列中mapping时,是将read或其子序列在按序排列的所有后缀中一一比对找到匹配位置。算法VCAKE、SSAKE、SHARCGS、BWA-SW应用前缀树数据结构,它将序列所有的前缀用树的结构表示,原理与后缀树相同。如图3所示为序列S=GATGAC的前缀树和前缀DAWG (DirectedAcyclic Word Graph,有向无环词图)。
由于后缀树结构在比对大型序列时,空间占用比较大,Abouelhoda等改进了Manber和My-ers提出的后缀树存储方法,称为增强后缀数组来提高空间利用率。Ferragina和Manzini提出的FM (full-text minute-space)-indexc是一种基于BWT (burrows-wheeler transform)的全文本压缩索引结构,利用BWT矩阵与后缀树之间的关系压缩后缀数组和索引,减少内存占用率。
第一步,BWT矩阵T设序列S以“$”为结束符号,轮换列出序列S所有的后缀记为数组M,然后将数组M的每行按照字母顺序排序得到BWT矩阵T;
第二步,矩阵T中第i行在矩阵M中的行号(也是矩阵T第一列字母在序列S中的序号)为数组S[i]矩阵T中最后一列的结果为BWrr数组B[i]。我们可以根据这两个数组将序列S还原,这个算法称为UNPERMUTE算法;
第三步,创建数组Oc (A)表示矩阵T中第一列的第一个A在第几行,Occ(2,G)表示矩阵T的最后一列中第2行的G是此列的第几个G。以这两个数组为索引可以很快找到匹配点;
用这种方法比对的软件有:Bowite、BWT-SW、BWA-SW。我们知道人类的基因组为3 Gb,那么数组Occ和S的每个值都要用4 Byte表示,3 Gb就要分别用12 GB内存存储。在Bowtie中应用了FM-index,每隔数行建立一个索引,使得人类基因组的内存占用约为1.3 GB,完全可以在普通机器上运行。
基于这两种数据结构的算法都有利于提高比对效率,两者的区别在于后缀树可以有效减少不精确匹配,并可避免比对过程中做的无用功,这个特点适用于相同物种之间相似性高的序列比对和寻找保守区。而应用哈希表数据结构的算法具有较高的敏感性,有利于发现SNPs和突变。可用于局部匹配或从大量数据巾搜索匹配点以及跨物种序列间的比对。
3 序列分析软件比较
表1中列出民目前常用的第二代测序序列的分析软件,对软件分析的序列类型、序列长度、软件特点、功能以及可输入、输出格式等方面进行了详细析和比较。
选择合适的软件要根据软件适用的序列类型、长度以及运用的算法、性能特点、输入/输出格式等项全而考虑,如表1中列出的常用软件的各项属性进行筛选:1)根据第2列中序列类型,如果是Spliced reads可以选择QPALMA和TopHat软件,(color-spaced reads可以选择BWT -SW、BFAST;2)根据第4列中read的长度,SSAHA、BLAT用于长序列的比对;SOAP.ZOOM、SeqMap适用于short-reads的mapping;3)按照表中5和6列软件的特点和功能,如Bowtie、BWT -SW、RMAP、MAQ、QPALMA、SHiRMP等在分析序列对,应用了质量分数,这在很大程度上提高了比对的准确性。用Bowlie和BWA在分析速度内存占用方面略优于其他软件:4)第8列为软件在比对时是否允许错配,gaps以及indels,这也影响比对结果。例如,MAQ在比对Illumina paired-end reads时允许gaps这样可以加大paired-end reads拼接的成功率。BWA、BFAST、SHiRMP、PASS等在进行比对时允许一定或不定数量的gaps可以提高比对的敏感度,降低假SNP的出现率,有利于发现真正的突变位点;5)最后,根据软件的输入格式是否符合待比对序列的格式、输出格式是否又符合下一步分析要求的格式进行软件选择。
此外,在分析数据的时候将两个或多个软件结合使用,例如在处理spliced reads时,可以使用Bowtie或BWT建立索引文件,再将索引文件使用TopHat、SoapSplice等软件进行splice junction检测。
4 结论
本文对国内外第二代测序技术以及数据的分析算法进行了总结和分析,对目前流行的序列分析软件进行详细归纳和比较并给出了参考建议,有利于从整体上把握测序技术和序列分析的发展状况.为今后序列分析和研究提供参考。
通过上述研究发现虽然基因分析软件已有很多且解决了一定的问题,但仍然存在困难:1)由于序列中大量repeat(重复片段)的存在,很多短reads作为repeat的一部分,不能对其准确定位,增大了拼装难度; 2)当前大多硬件环境较低,计算时间以小时或天为单位,严重限制了研究进展;3)比对结果的准确率和下一步分析的可用性方面有待提高。
针对上述问题,第三代单分子纳米孔测序技术目的是在第二代测序技术的基础上提高测序的准确性、增长read的读长。虽然其技术还不成熟,但必将成为未来几年的主流技术。序列比对方面,现有越来越多的软件提供并行处理方式,所以算法并行化以及云计算技术的应用将会成为序列分析的重要发展方向。
