定量计算已知易感变异对精神分裂症遗传度的解释度☆
2014-04-28李康许瑞环张洪德王前
李康 许瑞环 张洪德 王前
·论著·
定量计算已知易感变异对精神分裂症遗传度的解释度☆
李康*△许瑞环△张洪德△王前*
目的评估已发现精神分裂症易感变异对精神分裂症遗传度的解释程度。方法查询美国国家人类基因组研究所(National Human Genome Research Institute,NHGRI)编制的全基因组关联研究(genome-wide association study,GWAS)目录,检索所有已发现的精神分裂症易感变异位点共347个,纳入其中已提供风险等位基因频率和比值比的62个位点,使用多因素易患性阈值模型计算每个易感变异对精神分裂症遗传度的解释度。结果已知的62个精神分裂症易感变异对精神分裂症遗传度的合计解释度为25.66%,尚有74.34%的遗传度无法被已知易感变异解释,属于遗传度缺失。结论已知精神分裂症易感变异对精神分裂症遗传度的解释度依然较低,表明精神分裂症尚存在许多未知的分子遗传学机制,有待进一步阐明。
精神分裂症 全基因组关联研究 单核苷酸多态性
精神分裂症是典型的多基因遗传病,其发生受多个易感变异控制,遗传度为81%[1]。目前已有大量精神分裂症易感变异被发现,但已知的易感变异无法解释精神分裂症的高遗传度,存在“遗传度缺失(missing heritability)”[2]现象。2010年,So等[3]使用多因素易患性阈值模型[4],首次定量评估已知易感变异对包括精神分裂症在内的多种复杂疾病遗传度的解释程度,发现已知精神分裂症易感变异仅能够解释精神分裂症遗传度的0.39%。鉴于2010年至今,已有大量新的精神分裂症易感变异被发现,而国内外针对精神分裂症的遗传度解释度无进一步深入的报道,本研究拟收集目前所有已发现的精神分裂症易感变异,使用So等[3]的研究方法定量计算其对精神分裂症遗传度的解释度。
1 对象与方法
1.1 研究对象精神分裂症易感变异信息来源于美国国家人类基因组研究所(National Human Genome Research Institute,NHGRI)编制的全基因组关联研究(genome-wide association study,GWAS)目录(GWAS Catalog)[5](http://www.genome.gov/gwastudies)。截止2013年9月30日,目录中包含精神分裂症的易感变异位点347个,其中285个位点因未提供相应的风险等位基因频率(risk allele frequency)或比值比(odds ratio,OR)而被排除,本研究共纳入62个精神分裂症易感变异,详细情况见表1。
1.2 研究方法参考So等[3]基于多因素易患性阈值模型分析复杂疾病已知易感变异对疾病遗传度解释程度的方法进行分析。假设每个易感变异由一个风险等位基因A和一个保护等位基因a组成。对同一人群,三种基因型AA、Aa、aa具有不同的易患性平均值和相同的易患性阈值,如图1所示。A、a的等位基因频率分别为PA、Pa;AA、Aa、aa的基因型频率分别为PAA、PAa、Paa;AA、Aa、aa的外显率分别为fAA、fAa、faa。RR1为Aa相对于aa的相对风险度(relative risks,RR),RR2为AA相对于aa的相对风险度,本研究使用易感变异的OR值作为其RR1的估计值。基因型AA、Aa、aa的易患性平均值分别为μAA、μAa、μaa;T为疾病的易患性阈值;K为终生患病风险,精神分裂症的终生患病风险为0.0072[6]。
首先,根据Hardy-Weinberg平衡定律,可由PA推算Pa、PAA、PAa、Paa。K与基因型频率、基因型外显率、相对风险度的关系如下:
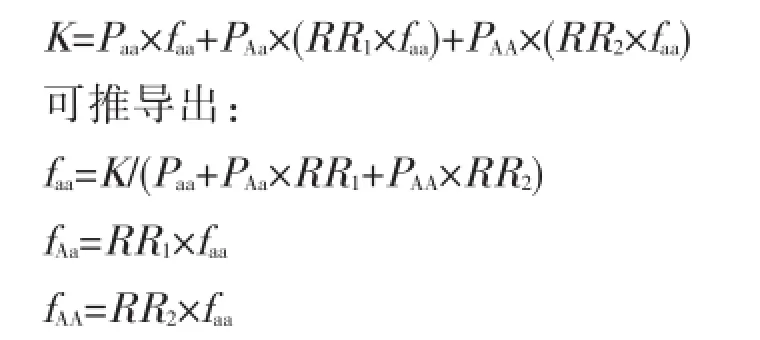
假设三种基因型的残余方差均为1,则基因型特异性易患性均值与基因型外显率存在如下关系:
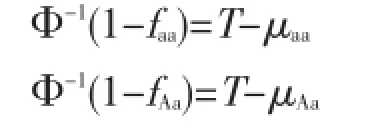
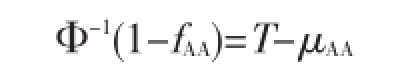
Φ-1为返回正态分布百分位数的函数。假设μaa数值为0,则T、μAa、μAA可分别计算如下:
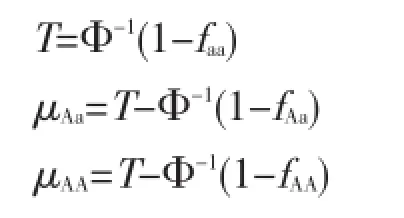
所有人群总易患性平均值μall计算公式如下:
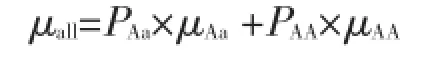
易患性解释变异(variance in liability explained,Vg)计算公式如下:

为方便与遗传度进行比较,将Vg按照如下公式进行变换,变换后的Vg称为实际Vg(actualVg)。

遗传度解释度=actualVg/h2h2为遗传度,精神分裂症的遗传度为81%[1]。使用R语言编写的相应软件(So等[3]提供http://sites.google.com/site/honcheongso/software/varexp)完成本计算。
2 结果
62个精神分裂症易感变异对于精神分裂症遗传度的合计解释度为25.66%,结果见表1。对精神分裂症遗传度的解释度高于1.00%的易感变异位点有7个,合计解释度为11.27%,其中解释度最高的是位于15q25.3的rs16977195,为3.55%。对精神分裂症遗传度的解释度低于1.00%的易感变异有55个,合计解释度为14.39%,其中解释度最低的是位于18q21.2的rs17512836,为0.03%。精神分裂症遗传度中尚存在74.34%无法被已知易感变异解释,为精神分裂症的遗传度缺失。

图1 易患性阈值模型示意图[3]μAA、μAa、μaa分别为基因型AA、Aa、aa的易患性平均值,阴影部分表示精神分裂症患者
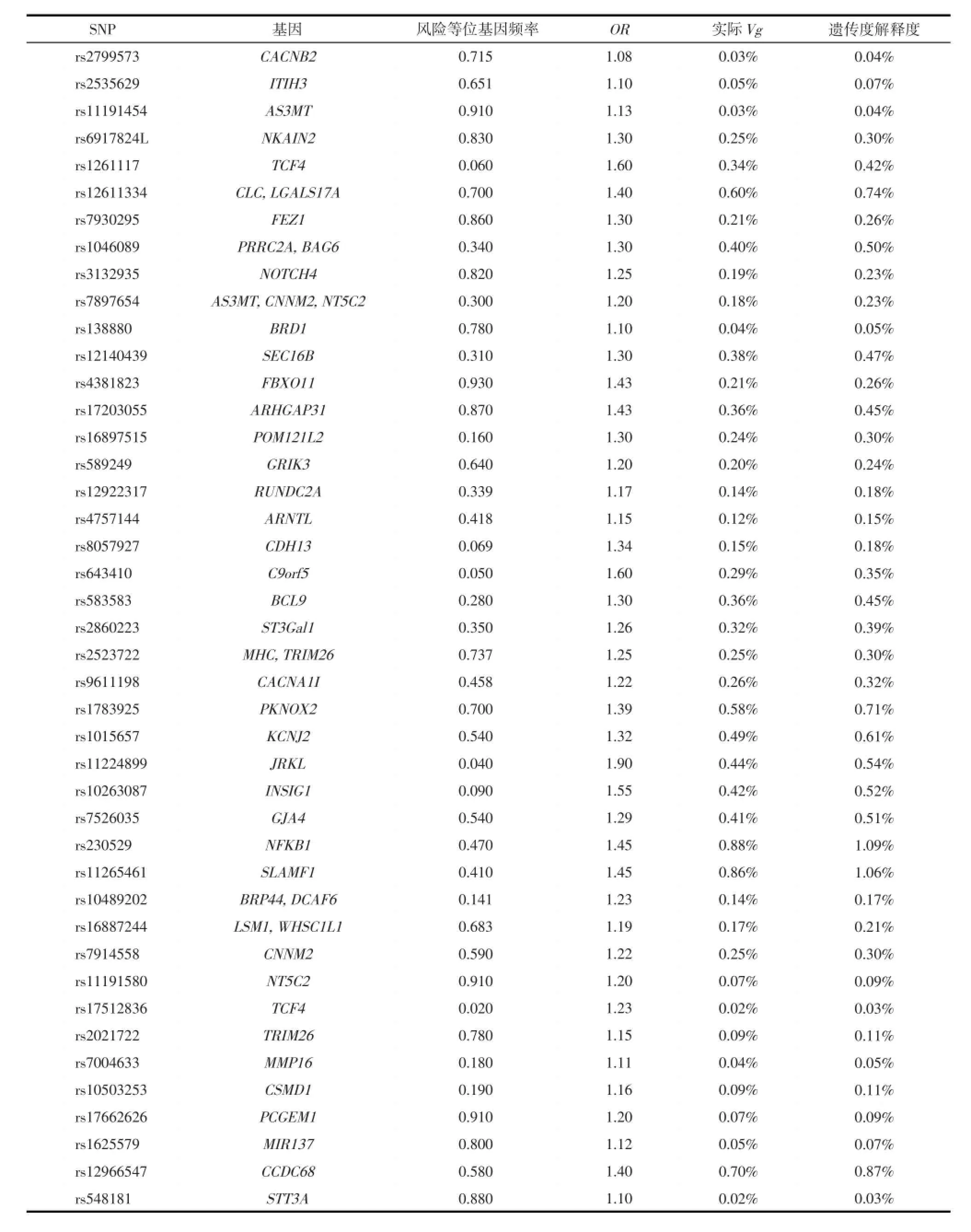
表1 已知精神分裂症易感变异对精神分裂症遗传度的解释度
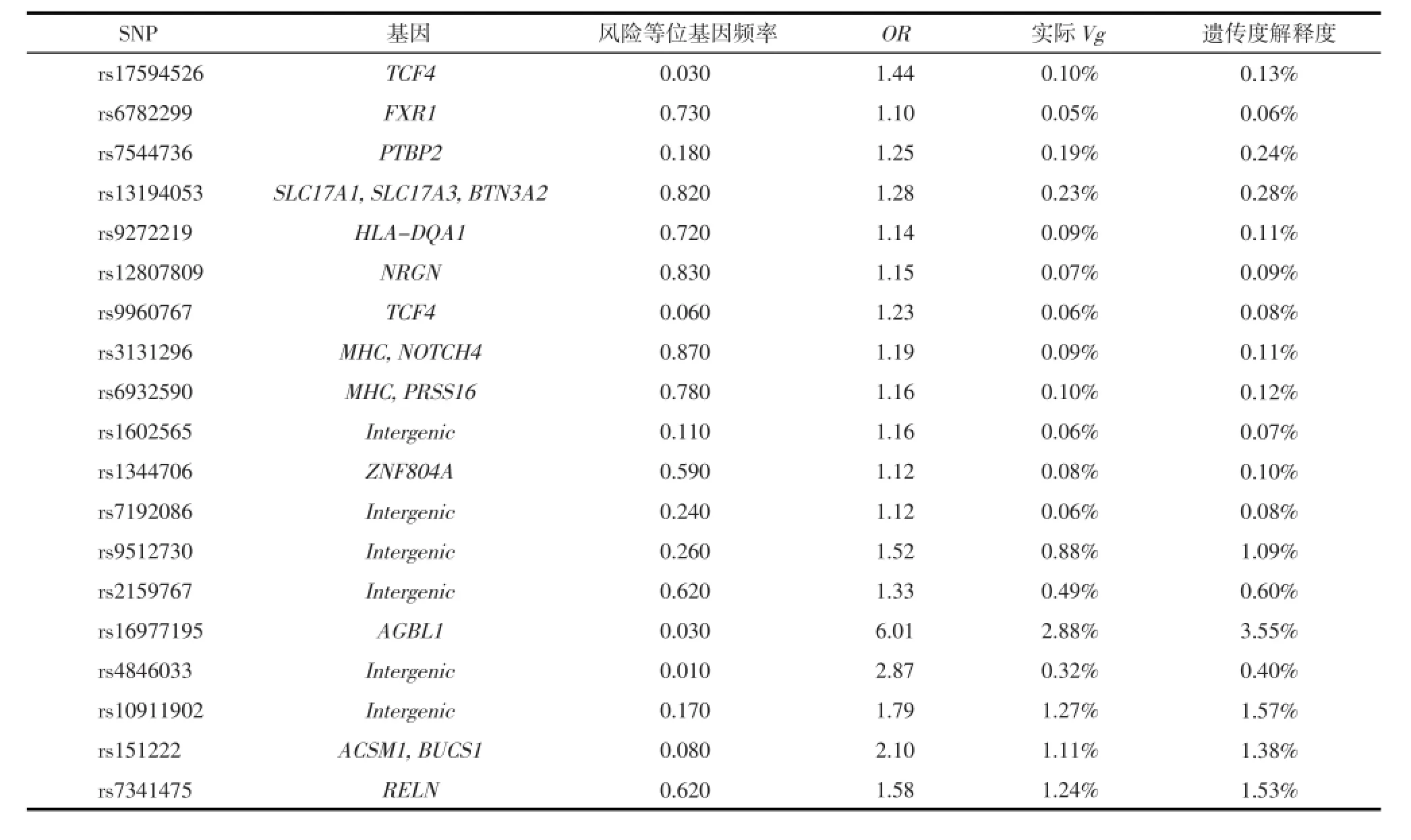
续表1
3 讨论
精神分裂症是典型的多基因遗传病,多对微效基因协同,并与环境因素共同作用导致疾病发生[7]。随着越来越多精神分裂症相关GWAS研究的开展,目前已发现超过300个精神分裂症易感变异。然而继续深入的研究显示已知精神分裂症易感变异无法完全解释疾病遗传度,存在遗传度缺失现象。
So等[3]对10种常见多基因遗传病的已知易感变异对遗传度的解释度进行定量计算,结果显示前列腺癌的解释度最高,为31.16%,解释度最低的精神分裂症仅0.39%。本研究发现精神分裂症易感变异对遗传度的解释度为25.66%,高于先前报道。分析其中原因,首先,纳入大量新发现的精神分裂症易感变异提高了本研究的总遗传度解释度。So等[3]的研究参考截至2010年1月31日的NHGRI GWAS目录,当时目录中仅收录33个精神分裂症易感变异,排除13个未提供风险等位基因频率或OR值的易感变异后剩余20个易感变异,So等[3]从这20个易感变异中选择4个,加上该目录外的4个,合计8个易感变异用于评估精神分裂症遗传度解释度。2010年2月1日至2013年9月30日,该目录新增214个精神分裂症易感变异,本研究共纳入62个,使已知精神分裂症易感变异对精神分裂症遗传度的解释度大大提高。其次,本研究未对NHGRI GWAS目录包含的所有精神分裂症相关GWAS研究设定统一的假设检验水准,未对已知精神分裂症易感变异进行二次筛选,使更多易感变异纳入本研究。So等[3]为保证结果准确度,使用7.2×10-8作为NHGRI GWAS目录中所有精神分裂症GWAS研究病例与对照间SNP频率比较的检验水准,对其结果进行二次筛选,其中13个精神分裂症易感变异被认为在病例、对照间缺乏统计学差异(P>7.2×10-8)而未纳入研究。考虑到各GWAS研究已根据自身样本量、研究SNP数量等因素选择了适合自身的检验水准,为尽可能全面地评估所有已发现精神分裂症易感变异对其遗传度的解释度,本研究未采用7.2×10-8作为统一检验水准进行二次筛选,从而使得更多的精神分裂症易感变异被纳入本研究。
本研究也存在一定的局限性。首先,资料来源于GWAS研究,而GWAS自身的局限性在一定程度上制约了本研究,如较难识别最小基因频率(minor allele frequency,MAF)小于5%的突变,以及对拷贝数变异(copy number variations,CNVs)和中性拷贝变异(copy neutral variation)等结构性变异较难识别等;第二,对于精神分裂症易感变异,本研究主要参考美国NHGRI编制的GWAS目录,可能存在一些易感变异由于未录入此目录而未被纳入本研究,从而使计算所得的精神分裂症遗传度解释度偏低;第三,本研究使用的计算方法要求易感变异必须有相应的风险等位基因频率和OR值,GWAS目录中与精神分裂症相关的易感变异总计347个,其中仅62个同时提供上述两个参数,即超过80%的易感变异由于缺少上述数据而无法纳入分析,这部分易感变异对精神分裂症遗传度的解释度无法评估;第四,本研究使用OR值近似替代RR值,但当疾病患病率较高时,OR值将大于RR值,使用OR值计算所得的遗传度解释度将大于使用RR值计算所得的结果。
已发现的精神分裂症易感变异仅能解释精神分裂症遗传度的25.66%,精神分裂症存在明显的遗传度缺失,这表明其遗传机制中仍有大量未知因素有待进一步研究。这些未知因素可能包括稀有突变的影响,拷贝数变异等结构性变异的影响,基因—基因、基因—环境相互作用的影响等。Manolio等[10]给出的复杂疾病遗传度缺失研究策略包括:对极端表型患者进行目标区域或全基因组测序,以尽可能发现与疾病相关的稀有突变及结构性变异;挖掘现有GWAS研究,研究基因结构变异与疾病的关联及基因间相互作用的证据;通过扩大单个研究和Meta分析来增加研究的样本量;增加对非欧裔人群的研究。相信随着全基因组外显子测序、全基因组测序等技术越来越广泛地应用于精神分裂症研究,研究样本量不断增大,基因—基因、基因—环境间相互作用不断阐明,以及对稀有变异和结构性变异与精神分裂症相关性的深入研究,精神分裂症的遗传机制将得到进一步阐明,其遗传度缺失也将不断缩小。
Lee等[11]建立了名为基因组复杂性状分析(ge nome-wide complex trait analysis,GCTA)的基于线性混合模型定量计算复杂疾病遗传度缺失的统计学方法,目前已被迅速应用于包括帕金森病[12]在内的众多复杂疾病遗传度缺失研究,并显示出良好的研究价值。我们计划在下一步的研究中使用GCTA方法对精神分裂症遗传度缺失进行进一步研究。
[1] Sullivan PF,Kendler KS,Meale MC,et al.Schizophrenia as a complex trait:evidence froma meta-analysis of twin studies[J]. Arch Gen Psychiatry,2003,60(12):1187-1192.
[2] Maher B.The case of the missing heritability[J].Nature,2008, 458(7218):18-21.
[3] So HC,Gui AH,Cherny SS,et al.Evaluating the heritability explained by known susceptibility variants:a survey of ten complex diseases[J].Genet Epidemiol,2011,35(5):310-317.
[4] Falconer DS.The inheritance of liability to certain diseases,estimated fromthe incidence among relatives[J].Ann HumGenet, 1965,29(1):51-76.
[5] Hindorff LA,Sethupathy P,Junkins HA,et al.Potential etiologic and functional implications of genome-wide association loci for human diseases and traits[J].Proc Natl Acad Sci USA,2009, 106(23):9362-9367.
[6] Bhugra D.The global prevalence of schizophrenia[J].PloS Med, 2005,2(5):e151.
[7] 闫景新,周玉萍,楚平华.精神分裂症的分子遗传学研究进展[J].中国神经精神疾病杂志,2009,35(1):60-64.
[8] Shifman S,Johannesson M,Bronstein M,et al.Genome-wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women[J].PloS Genet, 2008,4(2):e28.
[9] Sullivan PF,Lin D,Tzeng JY,et al.Genomewide association for schizophrenia in the CATIE study:result of stage 1[J].Mol Psychiatry,2009,14(12):1144.
[10] Manolio TA,Collins FS,Cox NJ,et al.Finding the missing heritability of complex diseases[J].Nature,2009,461(7265): 747-753.
[11] Lee SH,Wray NR,Goddard ME,et al.Estimating missing heritability for disease fromgenome-wide association studies[J].AmJ HumGenet,2011,88(3):294-305.
[12] Keller ME,Saad M,Bras J,et al.Using genome-wide complex trait analysis to quantify‘missing heritability’in Parkinson’s disease[J].HumMoi Genet,2012,21(22):4996-5009.
Quantitative evaluating the heritability explained by known susceptibility variants of schizophrenia.
LI Kang,XU Ruihuan,ZHANG Hongde,WANG Qian.Laboratory Medical Center,Nanfang Hospital,Southern Medical University,Guangzhou 510515,China.Tel:020-61641007.
ObjectiveTo evaluate the heritability explanation degree of schizophrenia by all known susceptibility variants in schizophrenia.MethodsThe GWAS catalog of National Human Genome Research Institute(NHGRI)was queried to retrieve all the susceptible gene variations of schizophrenia.Sixty-two variants with risk allele frequency and odds ratio(OR)were selected from347 susceptible gene variants of schizophrenia.The heritability exp lanation degree of each susceptibility variants was calculated using the multifactorial liability threshold model.ResultsThe total heritability explanation degree of schizophrenia by 62 known susceptible variants was 25.66%.In contrast,74.34%of heritability, which could not be explained by known susceptibility variants,were then defined as the missing heritability of schizophrenia.ConclusionsThe results demonstrate that the heritability explanation degree of schizophrenia by all known susceptibility variants in schizophrenia is low,indicating that there may be many unknown schizophrenia molecular genetic mechanisms need to be further clarified.
Schizophrenia Genome-wide association study Single nucleotide polymorphism
R749.3
A
2014-01-10)
(责任编辑:肖雅妮)
10.3936/j.issn.1002-0152.2014.08.001
☆广东省科技厅科技计划项目(编号:2011B061200003)
*南方医科大学南方医院检验医学中心(广州510515)△深圳市龙岗中心医院中心实验室
