聚合物在取向聚合物薄膜上的附生结晶研究进展
2014-04-13刘庆华
李 丽,刘庆华
(菏泽学院化学化工系,山东 菏泽274015)
0 前言
附生结晶是指结晶物质在取向基底上的结晶。附生结晶最早发现在天然矿物中,而系统研究开始于20世纪。Royer[1]在1928年首次提出附生结晶,早期的附生结晶研究主要集中在小分子体系。20世纪中期,半结晶聚合物在无机小分子基底上附生结晶现象的发现[2-5]拓展了附生结晶的研究领域,随后,半结晶聚合物在有机小分子基底[6-9]上的附生结晶研究也相继展开。20世纪80年代,聚合物/聚合物附生结晶才正式开始系统研究。在对聚合物/聚合物附生结晶的研究中,根据附生材料和基底的种类及其相互作用关系,聚合物/聚合物附生结晶可分为三大类,即均相附生结晶、异相附生结晶、拟态附生结晶。附生结晶的聚合物基底可分为取向基底和非取向基底,取向基底可通过熔体拉伸法、摩擦成膜法、附生制膜法制备,而附生聚合物的沉积方法有稀溶液结晶、熔体结晶、真空蒸涂气相结晶。目前,多从附生结晶的形貌,结晶动力学,结晶机理等方面进行研究。研究发现通过附生结晶可以控制不同聚合物的晶体结构和取向,如Lotz等[10-11]通过改变不同的基底成功控制了全同聚丁烯-1(i-PB)熔融结晶行为,从而得到几种不同晶型的i-PB。众所周知,材料的结构决定材料自身的性能,附生结晶时基底与附生聚合物界面间的物理结合作用使附生聚合物呈现不同的形态结构,从而导致材料的特殊性能及应用,如Lovinger[12]利用附生结晶从熔体中巧妙控制了生成具有压电和热电性功能的β相聚偏氟乙烯(PVDF)的方法。可见,聚合物附生结晶无论从理论上还是实践中都成为了重要的研究课题,因此本文总结了以取向聚合物膜为基底的聚合物/聚合物取向附生结晶的结晶机理及附生结晶的影响因素,为后续研究者提供参考基础。
1 附生结晶机理
聚合物/聚合物附生结晶分为成核和晶体成长2个过程。聚合物附生结晶是半结晶聚合物在取向聚合物界面上结晶,晶体是在二次核的基础上生长的,一次成核可不予考虑[13]。下面讨论聚合物附生结晶的二次成核过程,二次成核时对聚合物晶核长度有严格要求。首先,聚合物成核要求晶核达到临界晶核长度(l*)。根据经典 的Hoffman-Lauritzen 理 论[14],将 达 到临 界晶核长度的稳定晶核看做单一链杆,如图1,此时临界晶核长度与过冷度(ΔT,熔点与结晶温度的差)的关系可用式(1)表示:

式中 σe——折叠表面自由能
ΔHf——平衡熔点时的熔融热
从式(1)中可知过冷度一定时,只有形成长度为l*的晶核,晶体才能开始生长。实验证明上述公式对聚合物附生结晶仍然成立,只需做适当修正,相关修正工作目前仍在研究中。
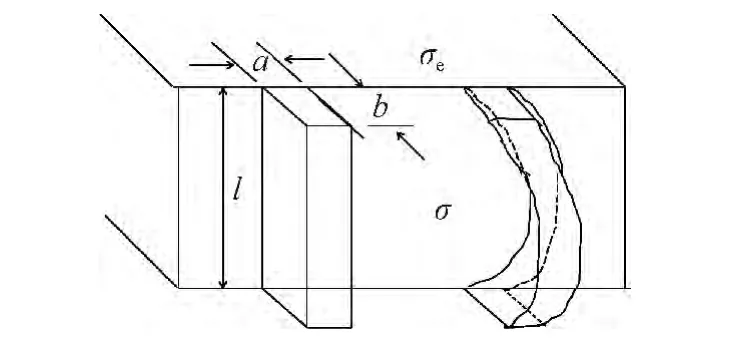
图1 附生结晶二次成核示意图Fig.1 Model for secondary nucleation
其次,聚合物成核要满足热力学公式的要求。聚合物沉积过程中会同时产生新的表面能(2blσ),并释放融化能(ablΔGf),二者部分抵消后剩余部分就是稳定晶核形成的能垒(Δφ1)。附生聚合物并非以伸展链方式沉积,而是部分有序沉积,如图1 右半部分所示,因此融化能在沉积过程中只释放一部分(ΨablΔGf),另一部分(Δφ2)在晶格定位时释放出来[15]。稳定晶核形成的能垒(Δφ1)的表达式为:
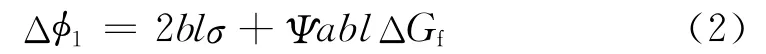
部分融化能(Δφ2)的表达式为:

式中 Ψ——融化能在沉积时的释放比率
ΔGf——沉积过程中单位体积释放的融化能
式中表面能大于零,沉积融化能小于零,即沉积放热,是聚合物结晶的动力。假设基底片晶的厚度为l′,则基底对沉积聚合物附生结晶方向的最大有效尺寸为:l′/cosθ,如图2。若l<l′/cosθ,附生聚合物链沉积,形成临界核;若l>l′/cosθ,沉积物链跨越了基底晶区和非晶区两部分,只有基底的晶区对沉积物起到诱导成核作用,因此非晶区的聚合物链对沉积物的融化能项无任何贡献,但对表面能项的贡献不变,式(2)变为:
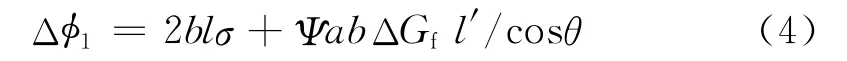
此时与正常结晶相比,附生结晶融化能项绝对值减少了ΨabΔGf(l-l′/cosθ),相当于结晶动力减小,导致附生结晶不能发生。
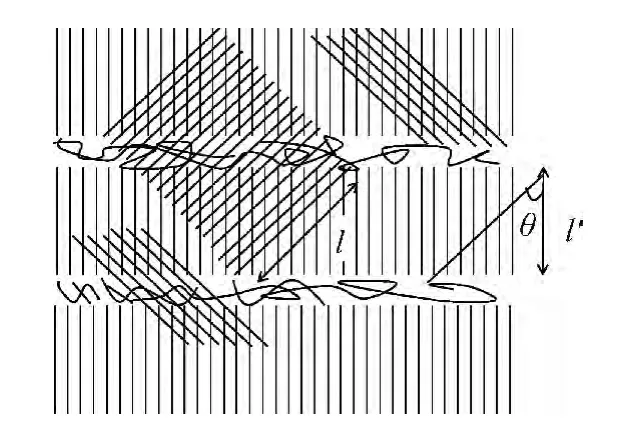
图2 附生结晶聚合物链沉积示意图Fig.2 Model for epitaxial crystallization chain deposited
因此聚合物附生结晶的沉积物片晶厚度必须介于临界晶核长度和基底对沉积聚合物附生结晶方向的最大有效尺寸之间才能形成稳定晶核。聚合物晶体生长过程可看作是聚合物多次的片晶折叠成核,附生结晶基底对沉积物有诱导成核作用,沉积物成核能力增强,整个过程的结晶速率会变快,尤其是对均相成核有困难的聚合物效果更明显,如等规聚甲基丙烯酸甲酯(i-PMMA)聚合物膜即使在最大结晶速率温度(120 ℃)下等温结晶一个月也很难看到明显的结晶现象,但以取向聚乙烯(PE)薄膜为基底附生结晶时,仅10h就可以完成结晶[16-17]。又如Tu等[18]研究发现聚乳酸(PLLA)自身结晶困难,但以取向PE 薄膜为基底附生结晶,即使在冷结晶的条件下,PLLA 也能取向结晶。此外,聚合物的膜厚、结晶温度等都会影响附生结晶的成核生长,我们将在下一部分详细的讨论。
2 附生结晶影响因素
附生结晶发生在基底与附生物的交界面上,与普通的聚合物结晶情况相比,影响因素更加复杂,掌握了附生结晶的影响因素可以更好地利用附生结晶改变聚合物的力学等性能,如Brinkmann[19-20]利用聚合物附生结晶的定向排列作用来改变半导体薄膜材料的性能,下面将对附生结晶的影响因素进行详细讨论。
2.1 晶格匹配
通常情况下,具有附生关系的2 种聚合物之间要求在结构上存在一维或者二维的匹配性,即结构相似。在聚合物附生研究中,附生结晶通常被认为是2种物质之间存在某种几何匹配,例如原子间距、分子间距、晶面间距等,即分子水平上存在相互作用,根据晶格匹配情况不同,聚合物异相附生体系可分为平行链和非平行链2种。为了研究匹配情况引入了失配率(Δ)的概念,Δ 的计算公式为:

式中 d、d0——附生物质、基底参与匹配的参数
一般认为附生结晶发生的失配率上限是10%~15%[21]。例如Lotz等[22-23]研 究PE 在 取 向 等 规 聚 丙烯(i-PP)基底上附生结晶,PE的(100)晶面内分子链间距为0.494,i-PP 在(101)方 向 的 甲 基 排 列 间 距 为0.505nm,失配率Δ 仅为2%,PE 与i-PP(101)方向的甲基晶格匹配,而i-PP(101)方向与i-PP分子链夹角为±50°,正好等于PE/i-PP附生结晶时两聚合物的分子链夹角。Liu等[16]研究发现i-PMMA 在高密度聚乙烯(PE-HD)基底上附生结晶时,二者的失配率仅为3.5%,i-PMMA 仅十几个小时就可以完成结晶,克服了自身结晶困难的问题。Sun等[24]对比研究了聚丁烯(PBA)在取向PE 基底、玻璃基底上的附生结晶,在玻璃基底上高温结晶时生成α 相晶体,低温结晶主要生成β相晶体,而以PE 作为基底时,由于β-PBA(a=0.506nm,b=0.735nm,c=1.467nm)与PE(a=0.74nm,b=0.494nm,c=0.2534nm)是相似的斜方晶系,PBA 在PE基底上附生结晶时只生成β-PBA,与PBA 的结晶温度无关。
附生结晶的界面分子间相互作用与物质种类有关。基底或沉积物不同,附生结晶晶格匹配不同,分子链的夹角不同,附生结构也就不同[25-26]。如PE在间规聚丙烯(s-PP)基底上附生结晶[26],s-PP(021)晶面的面间距为0.4465nm,PE(110)面内分子链间距为0.445nm,失配率为0.3%,s-PP(100)晶面上有凸起的{CH3,CH2,CH3}基 团,{CH3,CH2,CH3}基 团 排 在s-PP(021)方向,与s-PP 分子链方向成±37°,PE 在s-PP(100)晶面的附生结晶实际是PE 分子链沿{CH3,CH2,CH3}基团排列取向沉积的结果。PE/i-PP 附生结晶的分子链夹角是±50°,而PE/s-PP附生结晶的分子链夹角是±37°,可以看出晶格匹配对聚合物附生结晶结构的影响,表1给出了部分聚合物的附生结晶详细情况[23-24,27-30]。
聚合物结晶分为晶核形成和晶体生长2 个阶段,Yan等[31]研究了PE-HD 在取向i-PP膜与玻璃边界上的重结晶现象,i-PP基底上的PE-HD 形成相互交叉的附生片晶,i-PP 边 界 或 边 界 附 近 的i-PP 晶 体 诱 导 的PE-HD 取向晶核致使其晶体穿过i-PP基底边界,在玻璃上生长形成无规取向的杂乱片晶,从而得出结论:晶格匹配只影响附生结晶取向核的形成,晶核形成后结晶生长不受晶格匹配的影响,而是自发进行。

表1 聚合物附生体系Tab.1 Epitaxial crystallization system
沉积物与基底的晶格匹配是附生结晶产生的重要决定因素,但并不是唯一的决定因素。研究发现PE 的(110)晶面内的分子链间距为0.445nm,i-PP(010)晶面的甲基排列间距为0.425nm,二者之间存在良好匹配,但晶体生长有一个最快生长方向,PE 分子链在(100)面内的折叠优先于(110)晶面内的折叠,因此,PE(100)晶面在i-PP基底上附生结晶,而PE(110)晶面的附生结晶现象被抑制。事实证明晶格匹配并不能完全决定附生结晶的发生。
2.2 链段规整度
聚合物结晶是通过聚合物链的折叠堆砌实现的,聚合物的横向宽度不会是无限的,即附生结晶沉积物存在一个临界厚度,超过这一厚度,取向基底对附生物结晶影响力会急剧下降,不能产生附生结晶。沉积物附生结晶的临界厚度与其分子链的规整度有关[32-34]。研究证明沉积物链规整度好,附生结晶易发生,临界附生结晶层厚。当沉积物分子链规整度差,侧链比较多时,分子链的柔性会降低,而且形成晶体的结构会非常的松散,基底聚合物对其结晶的影响能力就变差。
为了进一步证明该结论,以PE-HD、线形低密度聚乙烯(PE-LLD)和低密度聚乙烯(PE-LD)作为研究对象,跟踪其在i-PP基底上的附生结晶现象[35]。PEHD 基本无支化,规整度最好,PE-LLD 含有部分分布比较均匀的短支链,PE-LD 高度支化,支链长短、分布不均匀,规整度最差,实验发现规整度最好的PE-HD在i-PP基底上最容易附生结晶,且150℃恒温10min,室温淬火,在i-PP基底上附生层厚度可达到250nm,而同样条件下的PE-LLD 最大附生厚度为120nm,链段规整度最差的PE-LD 的附生结晶较困难,最大附生层仅为30nm。实验中可以明显看出,基底条件不变的情况下,随着PE 分子链规整度的降低,其临界附生层厚度逐渐降低,附生结晶越来越难发生。
2.3 结晶动力学
依据晶格匹配原理,缓慢降温过程更有利于附生结晶,小分子化合物符合这一规律,而聚合物附生结晶恰好相反[36],缓慢降温反而有利于聚合物的附生结晶。通过研究降温速率对PE-HD 在i-PP基底的附生结晶发现,样品加热至150 ℃,恒温10min消除热历史,然后室温淬火后附生结晶厚度可达到250nm[35],而样品以0.5 ℃/min的降温速率降温结晶时,PE-HD的附生层厚度仅为120nm[31]。可见结晶降温速率低时,聚合物的附生结晶取向性已经变差。
根据时温等效性原理,降温速率慢,结晶时间长,等温结晶温度提高,低过冷度对附生结晶的影响是等效的,均不利于聚合物的附生结晶。比如Yan等[37]系统地研究了温度对PE-HD/i-PP 附生体系的影响,发现低温有利于附生结晶,而高温不利于附生,在结晶温度范围内即使温度差别很小,对附生结晶的影响也非常大。研究发现等温结晶温度低于120 ℃时,可以得到非常规整的附生结晶,当结晶温度提高到124 ℃时,PE-HD 的附生层厚度变薄,厚度仅为100nm,继续升高结晶温度至125 ℃,PE-HD 附生层厚度已经小于50nm,而当结晶温度高于125 ℃时,PE-HD 在取向的i-PP上已经不能附生结晶,呈无规排列。
2.4 二次成核
二次成核是附生结晶产生的苛刻控制因素,二次成核沉积物片晶厚度(l)必须小于基底聚合物在附生结晶方向的有效晶体尺寸(l′/cosθ),否则匹配再完美的聚合物也不能发生附生结晶,即基底聚合物的片晶厚度影响聚合物的附生结晶。这是因为二次成核过程中的稳定晶核形成的能垒可以用Δφ1=2blσ+ΨablΔGf表示[15],能垒越小越利于晶核的形成,式中融化能ΨablΔGf小于零,是附生结晶的动力,融化能越大越有利于结晶。当沉积物片晶厚度比基底的有效尺寸大时,沉积物跨越了基底的非晶区,非晶区的聚合物链对融化能没有任何贡献,相当于促进结晶的融化能的绝对值减少了ΨabΔGf(l-l′/cosθ),导致了形成稳定晶核的能垒Δφ1比沉积物正常结晶时的能垒大,所以沉积物的附生结晶不能发生。研究证明i-PP 做基底时,当片晶较厚(18nm)时,PE-HD 可在基底上形成完善的交叉附生结构,片晶较薄(13nm)时,PE-HD 熔融重结晶后在基底上无规排列,不能附生结晶。Chang等[28]研究了PCL在片晶厚度为30nm 的PE-HD 取向薄膜上的附生结晶,发现升温至80 ℃等温15min,然后室温结晶,PCL可以在取向PE-HD 薄膜上很好的附生结晶,得到片晶厚度6.5nm 的取向PCL,二者的分子链相互平行,PE-HD 晶体在匹配方向的尺寸(30/cos0°=30nm)完全满足大于PCL 在该结晶温度下的片晶厚度的条件,因此可以得到完善的附生结晶。
3 结语
附生结晶中聚合物和基底间的定向关系导致了聚合物晶体的各向异性特点,相应的物理和机械性能都有所改进,拓宽了聚合物材料的应用。目前研究人员对聚合物/聚合物附生结晶进行了一系列的实验,得到了一些基础理论成果,但对于附生结晶影响因素的定量描述方面还不是很清楚,同时附生结晶还面临着如何利用附生结晶设计聚合物形貌使其性能满足产品的特定要求的问题,在今后的发展中本领域的挑战仍是如何诱导附生结晶行为以制备先进的聚合物材料。如聚合物太阳能电池的效能非常低,但是让片状的供体和受体材料相互垂直的夹在两极之间可以提高电池的效能,因此可以通过设计器件中的聚合物结构得到高效光伏器件。
[1] Royer L.Experimental Research or Parallel Growth on Mutual Orientation of Crystals of Different Species[J].Bull Soc Fr Miner Cristallogr,1928,51:7-15.
[2] Tuinstra F,Baer E.Epitaxial Crystallization of Polyethylene on Graphite[J].J Polym Sci Polym Lett Ed,1970,8(12):861-865.
[3] Boucher E A.Growth of N-alkane Crystals on Graphite and on Carbon-fibre Surfaces[J].J Mater Sci,1973,8(1):146-148,119.
[4] Mauritz K A,Baer E J,Hopfinger A.Molecular Energetics of the Epitaxial Crytallization of Polyethylene on Alkali Halide Substrates[J].J Polym Sci Polym Phys Ed,1973,11(11):2185-2197.
[5] Ihn K J,Tsuji M,Isoda S,et al.Morphology of Cycloparaffins Crystallized Epitaxially on NaCl[J].Makromol Chem,1989,190(4):837-847.
[6] Macchi E M,Morossoff N,Morawetz H.Polymerrization in the Crystalline State.X.Solid-state Conversion of 6-aminocaproic Acid to Oriented Nylon 6[J].J Polym Sci Part A1,1968,6(8):2033-2049.
[7] Smith P,Pennings J.Eutectic Solidification of the Pseudo Binary System of Polyethylene and 1,2,4,5-tetrachlorobenzene[J].J Mater Sci,1976,11(8):1450-1458.
[8] Wittmann J C,Hodge A M,Lotz B.Epitaxial Crystallization of Polymers onto Benzoic Acid:Polyethylene and Paraffins,Aliphatic Polyesters,and Polyamides[J].J Polym Sci Polym Phys Ed,1983,21(12):2495-2509.
[9] Wittmann J C,Lotz B.Epitaxial Crystallization of Polymer on Organic and Polymeric Substrates[J].Prog Polym Sci,1990,15(6):909-948.
[10] Kopp S,Wittmann J C,Lotz B.Epitaxial Crystallization and Crystalline Polymorphism of Poly(1-butene):Forms III and II[J].Polymer,1994,35(5):908-915.
[11] Kopp S,Wittmann J C,Lotz B.Epitaxial Crystallization and Crystalline Polymorphism of Poly(1-butene):form I[J].Polymer,1994,35(5):916-924.
[12] Lovinger A J.Crystallization of theβPhase of Poly(vinylidene fluorid)from the Melt[J].Polymer,1981,22:412-413.
[13] Phillips P J.Polymer crystals[J].Rep Prog Phys,1990,53(5):549-604.
[14] Lauritzen J I,Hoffman J D.Theory of Formation of Polymer Crystals with Folded Chains in Dilute Solution[J].J Res Nat Bur Stds,1960,64A:73-102.
[15] Greso A J,Phillips P J.The Role of Secondary Nucleation in Epitaxial Growth:the Template Model[J].Polymer,1994,35(16):3373-3376.
[16] Liu J,Wang J,Li H,et al.Epitaxial Crystallization of Isotactic Poly(Methyl Methacrylate)on Highly Oriented Polyethylene[J].J Phys Chem B,2006,110(2):738-742.
[17] Brinkhuis R H G,Schouten A J.Thin-Film Behavior of Poly(methyl methacrylates).3.Epitaxial Crystallization in Thin Films of Isotactic Poly(methyl methacrylate)Using Crystalline Langmuir-Blodgett Layers[J].Macromolecules,1992,25(10):2717-2724.
[18] Tu C,Jiang S D,Li H H,et al.Origin of Epitaxial Cold Crystallization of Poly(l-lactic acid)on Highly Oriented Polyethylene Substrate[J].Macromolecules,2013,46(13):5215-5222.
[19] Brinkmann M,Rannou P.Effect of Molecular Weight on the Structure and Morphology of Oriented Thin Films of Regioregular Poly(3-hexylthiophene)Grown by Directional Epitaxial Solidification[J].Adv Funct Mater,2007,17(1):101-108.
[20] Kayunkid N,Uttiya S,Brinkmann M.Structural Model of Regioregular Poly(3-hexylthiophene)Obtained by Electron Diffraction Analysis[J].Macromolecules,2010,43(11):4961-4967.
[21] Wittmann J C,Lotz B.Epitaxial Crystallization of Polymers on Organic and Polymeric Substrates[J].Prog Polym Sci,1990,15(6):909-948.
[22] Lotz B,Wittmann J C.Tructural Relationships In Blends of Isotactic Polypropylene and Polymers with Aliphatic Sequences[J].J Polym Sci Polym Phys Ed,1986,24(7):1559-1575.
[23] Zhou H X,Yan S K.Can the Structures of Semicrystalline Polymers Be Controlled Using Interfacial Crystallographic Interactions[J].Macromolecular Chemistry and Physics,2013,214(6):639-653.
[24] Sun Y J,Li H H,Huang Y,et al.Epitaxial Crystallization of Poly(butylene adipate)on Highly Oriented Polyethylene Thin Film[J].Macromolecular,2005,38(7):2739-2743.
[25] Petermann Y,Xu Y,Loos J.Epitaxial Crystallization of Syndiotactic Polypropylene on Uniaxially Oriented Polyethylene[J].Polym Commun,1992,33(5):1096-1098.
[26] Schumacher M,Lovinger A J,Agarwal P,et al.Heteroepitaxy of Syndiotactic Polypropylene with Polyethylene and Homoepitaxy[J].Macromolecules,1994,27(23):6956-6962.
[27] Yan S,Katzenberg F,Petermann J,et al.A Novel Epitaxy of Isotactic Polypropylene(αphase)on PTFE and Organic Substrates[J].Polymer,2000,41 (7):2613-2625.
[28] Chang H B,Zhang J M,Li L,et al.A Study on the Epitaxial Ordering Process of the Polycaprolactone on the Highly Oriented Polyethylene Substrate[J].Macromolecules,2010,43(1):362-366.
[29] Yan C,Li H H,Zhang J M,et al.Surface-Induced Anisotropic Chain Ordering of Polycarprolactone on Oriented Polyethylene Substrate:Epitaxy and Soft Epitaxy[J].Macromolecules,2006,39(23):8041-8048.
[30] Trac A,Jeszka J K,Kucińska I,et al.Influence of the Crystallization Conditions on the Morphology of the Contact Layer of Polyethylene Crystallized on Graphite:Atomic force microscopy studies[J].J Appl Polym Sci,2002,86(6):1329-1336.
[31] Yan S,Petermann J,Yang D.Epitaxial Behavior of HDPE on the Boundary of Highly Oriented Ipp Substrates[J].Colloid Polym Sci,1995,273(9):842-847.
[32] Li H H,Yan S K.Surface-Induced Polymer Crystallization and the Resultant Structures and Morphologies[J].Macromolecules,2011,44(3):417-428.
[33] Loos J,Katzenberg F,Petermann J.Epitaxial Crystallization of Linear Low-density Polyethylene on High-density Polyethylene[J].J Mater Sci,1997,32(6):1551-1554.
[34] Zhou H X,Jiang S D,Yan S K.Epitaxial Crystallization of Poly(3-hexylthiophene)on a Highly Oriented Polyethylene Thin Film from Solution[J].J Phys Chem B,2011,115(46):13449-13454.
[35] Yan S K,Lin J,Yang D,et al.Critical Epitaxial Layers of Different Kinds of Polyethylene on Highly Oriented Isotactic Poly(propylene)Substrates[J].Macromo Chem Phys,1994,195(1):195-201.
[36] Wang J,Kaito A,Ohnishi S,et al.Temperature Effect on Epitaxial Growth of Poly(p-oxybenzoate)[J].J Macromol Sci B,1998,37(1):1-13.
[37] Yan S K,Yang D C.Critical Crystallization Temperature for the Occurrence of Epitaxy Between High-density Polyethylene and Isotactic polypropylene[J].J Appl Polym Sci,1997,66(10):2029-2034.
