系统性红斑狼疮合并Evans综合征临床特点
2014-04-09王来芳郑文洁曾小峰张奉春
王来芳,赵 清,王 立,郑文洁,赵 岩,曾小峰,张奉春
(中国医学科学院 北京协和医学院 北京协和医院风湿免疫科,风湿免疫病学教育部重点实验室,北京 100730)
ChinJAllergyClinImmunol,2014,8(3):170- 173
系统性红斑狼疮(systemic lupus erythematosus,SLE)是较为常见的自身免疫性疾病,多发生于育龄期妇女,可导致多系统多脏器损害。SLE患者血液系统受累并不少见,主要表现为白细胞减少、贫血、血小板减低,甚至出现全血细胞减少。Evans综合征为特发性血小板减少性紫癜(idiopathic thrombocytopenic purpura,ITP)和自身免疫性溶血性贫血(autoimmune hemolytic anemia,AIHA)先后或同时发生。Evans综合征患者常表现为迁延、反复发作的血小板减少及溶血,严重者可致死。目前,国内外对SLE合并Evans综合征的研究仅见少数报道,且多为个案。为提高临床对此认识,本文回顾性分析SLE合并Evans综合征患者特点,以期达到早期识别以利于改善预后。
资料和方法
研究资料
收集2004年1月至2012年7月北京协和医院风湿免疫科住院的SLE合并Evans综合征患者资料。SLE诊断符合美国风湿病学会1997年修正的SLE分类标准[1]。SLE疾病活动指数(systemic lupus erythematosus diease activity index,SLEDAI)评估:0~4分为病情稳定不活动,5~9分为轻度活动,10~14分为中度活动,>15分为重度活动[2]。Evans综合征符合ITP和AIHA的诊断标准[3],并经血涂片、骨髓涂片及活检证实,排除其他原因引起血液系统损害。
Evans综合征治疗效果标准
完全缓解(complete remission,CR):血小板升至100×109L,血红蛋白达120 gL且无溶血表现。部分缓解(partial remission,PR):血小板升至50×109L,血红蛋白达100 gL。无效:未达到CR和PR。复发:随访中达CR或PR的患者血小板降至50×109L以下和(或)血红蛋白低于100 gL并伴溶血表现。
研究方法
回顾性分析SLE合并Evans综合征患者临床表现,实验室检查,治疗及预后等临床资料。
统计学处理
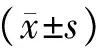
结 果
一般情况
共纳入SLE合并Evans综合征患者22例,占同期住院成人SLE患者的0.65%(223400例)。其中,男3例,女19例,年龄16~53岁,平均年龄(35.1±11.5)岁。22例患者中11例(50%)以血液系统损害为首发表现,从首发血液系统损害至确诊SLE的时间为6个月~18年,中位时间为124个月。其中,6例以ITP首发,男1例,女5例,平均年龄(40.3±10.6)岁,首发症状至确诊间隔中位时间为114个月;5例以AIHA首发,男1例,女4例,平均年龄(29.6±7.1)岁,首发症状至确诊间隔中位时间为48.5个月。
6例患者在SLE确诊后4~132个月后出现Evans综合征,中位时间68个月;这6例中男1例,女5例,平均年龄(35.5±16.8)岁。11例患者先确诊Evans综合征,至确诊SLE的间隔中位时间为26个月;这11例患者中,男2例,女9例,平均年龄(32.6±10.9)岁。二者同时诊断者5例,均为女性,平均年龄(38.0±1.8)岁。伴发其他结缔组织病5例(22.7%),抗磷脂抗体综合征3例,系统性硬化症和原发性胆汁淤积性肝硬化各1例。其中1例以血栓首诊为抗磷脂抗体综合征患者于确诊SLE间隔10年。
临床表现
所有22例患者中,肾脏受累13例(59.1%)(肾病综合征6例,肾功能不全1例),皮肤黏膜受累9例(40.9%)(脱发2例、光过敏6例、口腔溃疡1例),关节炎9例(40.9%),面部红斑6例(27.3%),神经系统受累4例(18.2%),雷诺现象、消化系统受累、肺部受累各2例(9.1%),心脏受累和浆膜腔积液各1例(4.5%)。SLEDAI评分3~30分,平均(11.5±7.6)分。SLE重度活动5例,中度活动4例,轻度活动10例,病情稳定3例。
实验室检查
所有22例患者均出现贫血、血小板减少。血红蛋白最低值为26~102 gL,平均61 gL;其中,中度贫血(60~90 gL)11例,重度贫血(30~60 gL)9例,极重度贫血(<30 gL)2例。血小板计数最低值为1×109L~90×109L,平均58×109L,其中20×109L~50×109L者8例,<20×109L者6例。15例患者直接Coombs试验阳性,2例患者Coombs试验阴性但有明显的临床溶血证据。超声检查示3例肝脏肿大,14例脾脏肿大,1例肝、脾均肿大。
22例患者血清抗核抗体均阳性,抗双链DNA抗体阳性6例(27.3%),抗SSA抗体阳性7例(31.8%),抗Sm抗体阳性4例(18.2%),抗核糖核蛋白抗体阳性5例(22.7%),抗核糖体核糖核蛋白抗体阳性6例(27.3%),抗磷脂抗体阳性6例(27.3%)(抗心磷脂抗体阳性6例,狼疮抗凝物阳性4例,抗β2糖蛋白1抗体阳性2例),抗血小板抗体阳性3例(13.6%)。补体下降17例(72.3%),免疫球蛋白升高13例(59.1%)。
治疗及转归
转归:21例患者有不同程度好转,13例达到CR,8例PR。其中1例经过多次甲泼尼龙冲击、静脉人免疫球蛋白及长期大剂量激素联用多种免疫抑制剂(环磷酰胺长春新碱环孢素)治疗,病情反复复发(血红蛋白40~50 gL),并出现不良反应(股骨头坏死和高血压),应用利妥昔单抗375 mgm2,3次,达到CR。随诊12~24个月,11例病情反复,多为糖皮质激素减至5~15 mgd时复发,再次增加激素剂量并联合多种免疫抑制剂后好转。1例死于狼疮肾衰竭合并原发性胆汁淤积性肝硬化所致肝硬化、肝性脑病。
讨 论
血液学异常表现是SLE患者最常见的症状之一,几乎所有SLE患者在病程中均会出现1种或几种血液学异常表现,部分SLE患者以血液系统损害为首发表现,甚至可在SLE典型症状前多年出现,早期易被误诊为血液病。Evans综合征罕见,约5%~10%的SLE患者可伴AIHA,伴血小板减少的SLE约占20%~40%[3- 4]。Evans综合征合并SLE罕见,以Evans综合征起病者约3%~15%可发展为SLE[5-6]。
本研究发现,SLE合并Evans综合征患者50%以血液系统(ITPAIHA)为首发表现,某些患者较长时间仅有血液系统表现,而无其他系统受累,自身抗体阴性,或低滴度抗体阳性,但随着病程发展,抗体逐渐出现或滴度增高,并出现典型的SLE特异性抗体如抗Sm抗体及抗双链DNA抗体阳性。因此,早期不易引起重视,导致发病时间与诊断时间间隔较长,仅有血栓形成者更易被忽视,本文1例以血栓首诊为抗磷脂抗体综合征患者,间隔10年才确诊为SLE。
SLE伴发Evans综合征以女性多见,且Evans综合征多出现在SLE多系统性损害及疾病活动时,临床易并发口腔溃疡、浆膜腔积液和肾脏损害[6]等。本组患者亦以女性为主(男女之比为3∶19)。SLE伴发Evans综合征时,患者常有多系统受累,发病率最高为肾脏受累(59.1%),其次是皮肤黏膜和关节炎(40.9%)、神经系统受累(18.2%)、胃肠道和肺部(9.1%)。另外,本组5例SLE伴发Evans综合征患者同时还合并其他自身免疫性疾病,与文献[6]相符。
SLE患者伴发Evans综合征的发病机制尚不十分清楚。目前认为,二者均为自身免疫疾病,主要与免疫介导有关,体内存在针对红细胞和血小板的自身抗体的免疫系统。该类患者体液免疫和细胞免疫同时存在,体液免疫作用可能更大,表现为T细胞功能紊乱,B细胞产生针对骨髓血细胞的自身抗体增多,多种免疫复合物和抗体存在血液中,免疫监视和自身识别功能发生障碍,使血细胞及骨髓受到破坏。研究显示,SLE伴发AIHA和(或)ITP患者抗磷脂抗体(包括抗心磷脂抗体和抗β2糖蛋白Ⅰ抗体)升高,可能参与了致病[3,7-8]。另有研究发现,Evans综合征患者病情活动时CD4+CD25+调节性T细胞(regulatory T cell,Treg)显著降低,随着病情缓解而升高,因此CD4+CD25+ Treg免疫调节异常可能与Evans发病和预后有关[9-10]。
Evans综合征患者多对糖皮质激素反应良好,但与ITP和AIHA不同,Evans综合征病情迁延、不易彻底治愈,激素减药过程中容易复发。目前尚没有明确的治疗指南,初始治疗仍以糖皮质激素和(或)免疫球蛋白为主。难治型患者可考虑行切脾手术、应用达那唑、免疫抑制剂、血浆置换等方式。利妥昔单抗为抗CD20单克隆抗体,能特异性杀伤表达CD20的B细胞,已应用于多种自身免疫病的治疗。同时,利妥昔单抗已成为ITPAIHA标准二线治疗药物,以减少脾脏切除率。近年来,国际上用利妥昔单抗治疗部分难治性和复发性Evans综合征患者也取得了一定疗效[11-12]。本研究所有患者均应用糖皮质激素治疗,其中12例患者进行了激素冲击治疗,且所有患者均应用1种或以上免疫抑制剂。然而,不论哪种治疗方法,患者疾病复发率仍较高。本文研究发现,糖皮质激素减至15 mgd是疾病复发的高峰,联合应用多种免疫抑制剂可更好使糖皮质激素减量,而在应用免疫抑制剂控制SLE其他症状同时也使Evans症状得以控制或有助于延长疾病缓解期,减少复发。
综上所述,SLE合并Evans综合征罕见,发生于SLE多系统受累及活动期。部分患者在SLE确诊前多年表现为ITP或AIHA,早期自身抗体不特异,症状不典型,易被误诊为血液系统疾病而延误治疗。因此,对于存在血细胞减少尤其是ITP和(或)AIHA患者,应详细追问病史,全面查体,及时筛查多种自身抗体,定期随访密切观察,以期早期诊断和治疗。
[1]Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus[J].Arthritis Rheum, 1997, 40:1725.
[2]Bombardier C,GIadman DD,Urowitz MB,et al.Derivation of the SLEDAI:a disease activity index for lupus patients:the committee on prognosis studies in SLE[J].Arthritis Rheum,1992,35:630-640.
[3]Kokori SI, Ioannidis JP, Voulgarelis M, et al. Autoimmune haemolytic anaemia in patients with systemic lupus erythematosus[J].Am J Med, 2000,108:198-204.
[4]Durán S, Apte M, Alarcón GS, et al. Features associated with, and the impact of, hemolytic anemia in patients with systemic lupus erythematosus:LX, results from a multiethnic cohort[J].Arthritis Rheum, 2008,59:1332-1340.
[5]Karpatkin S. Autoimmune thrombocytopenic purpura[J].Blood, 1980, 56:329-343.
[6]Costallat GL, Appenzeller S, Costallat LT. Evans syndrome and systemic lupus erythematosus:clinical presentation and outcome[J].Joint Bone Spine, 2012, 79:362-364.
[7]Sultan SM, Begum S, Isenberg DA. Prevalence, patterns of disease and outcome in patients with systemic lupus erythematosus who develop severe haematological problems[J].Rheumatology (Oxford), 2003,42:230-234.
[8]曾晓红,薛原. 抗β2糖蛋白Ⅰ抗体在系统性红斑狼疮合并自身免疫性溶血性贫血中的意义[J].中华临床免疫与变态反应杂志,2014, 8:28-34.
[9] Giovannetti A, Pierdominici M,Esposito A,et al.Progressive derangement of the T cell compartment in a case of Evans syndrome[J].Int Arch Allergy Immunol,2008,145:258-267.
[10] Liu B, Zhao H, Poon MC, et al. Abnormality of CD4 (+) CD25 (+) regulatory T cells in Idiopathic thrombocytopenic purpura[J].Eur J Haematol, 2007, 78:139-143.
[11] Tamimoto Y, Horiuchi T, Tsukamoto H, et al. Harada M. A dose-escalation study of rituximab for treatment of systemic lupus erythematosus and Evans’syndrome:immunological analysis of B cells, T cells and cytokines[J].Rheumatology, 2008, 47:821-827.
[12] Kittaka K, Dobashi H, Baba N, et al. A case of Evans syndrome combined with systemic lupus erythematosus successfully treated with rituximab[J].Scand J Rheumatol, 2008, 37:390-393.
