系统性硬化症动物模型
2014-04-09罗妍,王迁
罗 妍,王 迁
(中国医学科学院 北京协和医学院 北京协和医院风湿免疫科,风湿免疫病学教育部重点实验室,北京 100730)
ChinJAllergyClinImmunol,2014,8(3):248- 253
系统性硬化症(systemic sclerosis,SSc)是一种以皮肤及多系统炎性纤维化、小血管病变为主要特征的自身免疫疾病。其发病机制不清。目前认为,包括血管病变、炎性反应、纤维化及自身免疫4大机制相互作用。血管病变可能是始动因素,血管内皮细胞凋亡或功能紊乱,毛细血管舒缩功能、通透性改变,之后血栓形成、炎性反应激活,最终导致血管闭塞、组织缺血缺氧。炎性反应发生于疾病早期,皮肤病变以CD4+T细胞及单核细胞浸润为主,而肺部病变处则以CD8+T细胞为主,二者可能发病机制不完全相同。患者血清发现多种自身抗体,其中最具有标志性的是抗DNA拓扑异构酶Ⅰ抗体(抗Scl-70抗体)及抗着丝点抗体。处于核心地位的纤维化主要由血管病变和炎性反应起始,多种生长因子、趋化因子参与调控,转化生长因子(transforming growth factor,TGF)β通路主要参与,最终促进纤维化过程[1]。
对SSc发病机制的了解有助于及早诊断和探索更有效的治疗手段,因此能够模拟疾病的动物模型起到不可或缺的作用。目前,已有多种模拟SSc的动物模型,但无一种能完美代表这种疾病,不同的研究目的需选择不同的动物模型,本文将就目前常用的SSc动物模型的建模方法、模拟性及应用方面进行综述,希望能为研究者选择正确的、合适的动物模型提供帮助。
动物模型分类
SSc的动物模型从建模方法上可以分为两大类,即诱导模型和基因模型。前者主要包括博来霉素诱导小鼠模型、慢性硬皮病样移植物抗宿主病模型及活性氧簇活性氧离子(reactive oxygen species,ROS)介导模型;后者则又分为突变模型及转基因模型,突变模型主要包括紧皮鼠(tight skin mouse,TSK)模型和UCD200206小鸡模型,转基因模型则包括Fra-2、TBRICA; Cre-ER、TβRIIΔk、Caveolin-1模型等。前文简述了SSc的4大基本特点,故将主要从这些方面将模型与SSc患者进行比较(表1)。
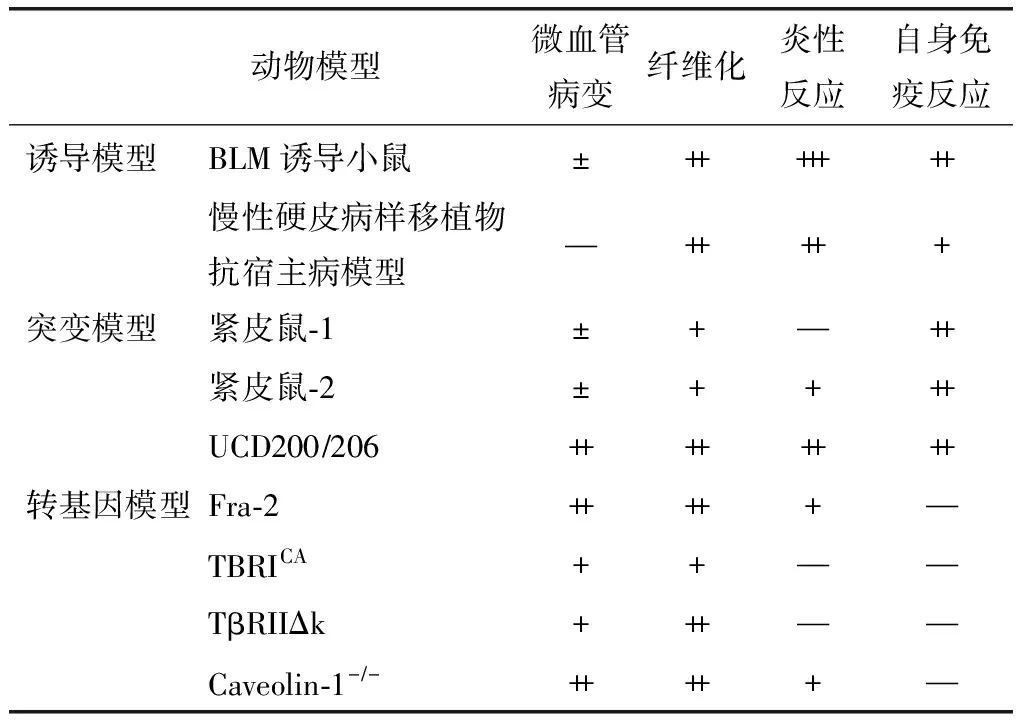
表1 常见动物模型主要特点
诱导模型
博来霉素诱导小鼠模型:该是目前应用最广泛的一种硬皮病动物模型,它可模拟硬皮病早期炎性反应阶段[2]。早期常用博来霉素给小鼠气管内灌注或吸入建立肺纤维化的动物模型[3],后发现皮下注射数周后可出现注射局部皮肤纤维化、肺纤维化及血清抗体阳性等SSc表现[3-5]。不同品系小鼠敏感性不同,BALBc小鼠相对耐药,而C3HHe、B10.A等更加敏感[5]。该模型小鼠纤维化部位可见胶原沉积、TGFβ1转录增加、肌成纤维细胞分化激活,并伴以巨噬细胞、T细胞及浆细胞为主的炎性细胞浸润[6],辅助性T细胞(helper T cell,Th)2反应相关因子白细胞介素(interleukin,IL)- 4、IL-13水平升高[5,7]。小鼠血清抗核抗体(anti-nuclear antibody,ANA)(+),包括抗Scl-70抗体、抗CENP B抗体、抗U1RNP抗体、抗组蛋白抗体、抗双链DNA抗体等[7]。该模型总体毛细血管含量增加不符合SSc患者特点,也未见硬皮病特征性的中小动脉闭塞性血管病[3]。其发病机制可能由活性氧簇ROS介导免疫炎性反应,最终激活成纤维细胞及TGFβ通路,引起纤维化[7](图1)。
博来霉素诱导小鼠模型广泛应用于SSc发病机制研究及新药治疗效果观察。目前研究以基因层面、蛋白质水平、细胞水平较多,发病机制方面与免疫炎性反应有关的靶点包括T细胞、B细胞、炎性细胞与成纤维细胞间的相互作用以及相关细胞因子如IL-6[8]、IL-17[9]、IL-33[10]、TNF-α[11]等,纤维化通路靶点则从经典的TGFβSmad通路[7]、Wnt通路[12]中参与信号转导的各种蛋白,到新发现的TGFβ非Smad通路[7]、黏附分子[13]以及某些激素等[14]。转录翻译的调控层面例如表观遗传学、miRNA的研究相对较少。治疗探索方面,与发病机制相关的有过氧化物酶体增生物激活受体[15]、酪氨酸激酶[16]、TGFβSmad通路,其他还有N-乙酰半胱氨酸[17]、雷帕霉素[18]、沙利度胺[19]等,甚至干细胞治疗[20]、皮肤光照治疗PUVA[21]等。但该模型炎性反应过重且非炎性反应的后期阶段表现相对较弱,某些情况下尤其是治疗学研究方面必须与非炎性模型联合应用[2]。另外由于几乎不能模拟血管病变,故在血管病变的研究受到限制。
慢性硬皮病样移植物抗宿主病模型:慢性硬皮病样移植物抗宿主病模型(chronic sclerodermatous GVHD model),以下简称Scl-GVHD模型,也是一种以炎性表现为特点的动物模型。该模型的建立基于进行过异体造血干细胞移植的长期生存者中约40-60%会出现自身免疫病表现的慢性移植物抗宿主病[3]。1983年Jaffee等将B10.D2小鼠的胫骨、股骨骨髓及脾制成淋巴细胞悬液移植入接受亚致死剂量照射过的BALBc小鼠体内,3周左右受体小鼠出现全身多器官纤维化[22]。如将受体换成固有免疫缺陷的RAG-2小鼠可降低死亡率,其肺纤维化相对较轻而皮肤纤维化更严重[23]。标准Scl-GVHD模型在移植后3周左右出现肺、皮肤、肝、肾、胃肠道、腮腺等多器官纤维化,在纤维化出现之前1周病灶处有以单核细胞为主的炎性细胞浸润,且这些细胞为供体来源[3]。小鼠血清ANA(+)但种类不明。其机制尚不明,目前认为小鼠血清及组织中趋化因子升高可能参与炎性反应及纤维化。Scl-GVHD因移植技术比较复杂而应用受到一定限制,且移植后动物处于免疫抑制易发生感染而死亡,故应用相对较少[3]。

图1 博来霉素诱导模型机制Fig 1 Mechanism of bleomycin-induced model ECM:细胞外基质
ROS介导的动物模型:既往研究发现,SSc患者的成纤维细胞可自发产生大量活性氧簇ROS,可促进胶原合成[7],在此基础上2009年建立了ROS介导的小鼠模型。给小鼠皮下注射过氧亚硝酸盐(ONOO-)可引起注射部位限制性皮肤损害以及血清抗CENP B抗体阳性;如皮下注射次氯酸(OH·HOCl)持续6周则可引起小鼠皮下及肺纤维化、肾损害以及血清抗Scl-70抗体(+)。这两种试剂注入体内后均可产生ROS。该模型病灶局部有T细胞为主的炎性细胞浸润,早期可见到血管内皮细胞破坏[7,24]。该模型由于建立时间较短而应用不多。研究者利用该模型发现SSc皮肤及肺部纤维化发病机制不同的可能机理,皮肤原位为成纤维细胞内形成ROS直接激活Ras通路,而ROS可氧化的蛋白可入循环诱导自身抗体产生,并到远隔脏器引起炎性及纤维化。在该模型中首次证实抗Scl-70抗体直接参与致病(图2)。
基因突变模型
TSK模型:TSK模型包括TSK-1及TSK-2,其基因突变位点不同但表现相似。B10.D2(58N)Sn近交系小鼠2号染色体原纤维蛋白基因1(fibrillin gene 1,Fbn-1)发生显性突变,纯合体在胚胎阶段死亡,杂合体就是TSK-1小鼠模型,该基因可编码细胞外基质的结构蛋白原纤维蛋白,故该模型的最显著特点是纤维化[25]。TSK-1小鼠具有皮肤和肺纤维化,但其病理特点与SSc患者并不完全相符合:小鼠皮肤纤维化发生于皮下、较深,筋膜层、肌层等受累,而SSc患者皮肤纤维化部位多位于真皮内;小鼠肺部受累以肺气肿为主要表现,在SSc患者中无此特点[26]。TSK-1小鼠皮肤病灶处无炎性细胞浸润的证据,故为非炎性模型的代表。未见小鼠发生内皮凋亡、小动脉闭塞等其他血管病变。该模型血清ANA升高,鉴定出的类型有抗Scl-70抗体、抗双链DNA抗体、抗RNA聚合酶Ⅰ抗体等[3]。TSK-1模型常与博来霉素诱导模型联合应用,后者主要模拟早期炎性阶段,而该模型主要用于模拟纤维化阶段。就目前已发表的研究来看,该模型的独特之处在于发现明确的B细胞活化,故较多应用于B细胞如何参与发病机制以及以B细胞为目标的治疗探索。但该模型纤维化病变与系统性硬化患者不完全相符合,应用受到一定限制。TSK-2小鼠为101H小鼠由致突变剂乙基亚硝基诱变后的子代,亦为常染色体显性突变,尚未确定责任基因。该模型与TSK-1相比具有以单核细胞浸润为主的炎性反应,其余表现类似,比TSK-1在模拟性方面更加全面[27]。目前因机制不清,应用较少。
转基因模型
转基因模型由于依赖较为复杂的转基因技术,并且靶点单一、无法模拟疾病全貌,故应用相对不是特别广泛,在这里对一些模型做简要介绍。
Fra-2是转录因子激活蛋白(activator protein-1, AP-1)家族成员,参与应激反应、细胞增殖、凋亡、炎性反应、损伤修复、癌变等许多生命基本过程。2008年偶然在由H2KB启动子介导的Fra-2过表达小鼠中观察到肺纤维化[30],后又发现其具有微血管内皮凋亡、皮肤毛细血管减少、肺动脉高压等血管病变[31]。肺动脉高压会使小鼠寿命极大降低,影响其应用。由于目前只有极少数模型能够模拟系统性硬化的血管病变,尤其是SSc相关肺动脉高压,故在未来的研究中具有一定的意义[2-3]。
TGFβ通路是纤维化中一条非常重要的通路,因此通过持续激活该通路可能创造新的纤维化模型。如TBRICA; Cre-ER转基因小鼠是在TGFβRI基因的上游引入增强子和他莫昔芬诱导Crelox P系统,小鼠出生后通过注射他莫昔芬来持续性激活成纤维细胞内TGFβRI表达[32]。小鼠出现皮肤纤维化,不会自发出现肺纤维化,但对博来霉素比普通小鼠更加敏感。病灶无明显炎性浸润,无典型血管病变,血清未见自身抗体。另一种TβRIIΔk转基因小鼠的目标基因是一种激酶有缺陷的突变TGFβRII,突变后该受体可结合胞外游离的TGFβ,但随后不能磷酸化TGFβRI并进一步起始信号转导,使得体内TGFβ代偿性上调,TGFβ通路被激活[33]。这种小鼠在生后6周出现肺纤维化,12周时约25%的小鼠出现皮肤纤维化,另外部分还可出现纤维性心肌病[3]。这些模型也主要应用于TGFβ通路介导的纤维化的研究。
Caveolin-1是一种特异性脂筏,可介导TGFβ受体内化而使下调TGFβ通路,该蛋白敲除的小鼠以纤维化病变和血管病变为主[34]。小鼠可出现肺部和皮肤纤维化,其肺部临床类似于放射性肺炎,病理表现为肺泡间隔细胞外基质蛋白沉积。小鼠微血管病变表现为血管因内皮细胞功能紊乱引起张力异常、通透性增加,大小动脉均可受累,并可出现肺动脉高压。该模型机制比较复杂,因为Caveolin-1作为脂筏可引起除TGFβR之外的其他受体内化而影响其他多个通路。但模型本身却为SSc的治疗提供一种思路,即通过外源引入Caveolin-1来促进TGFβ通路下调从而抑制纤维化[35]。
总 结
SSc的动物模型中,能够模拟早期炎性阶段的主要模型为博来霉素诱导模型、慢性硬皮病样GVHD模型,以纤维化为主要特点的模型有TSK模型、各种以TGFβ通路为靶点的转基因模型等,而进行微血管病变的相关研究则主要利用突变模型UCD200小鸡以及转基因模型Fra-2小鼠。在这些所有模型中,没有一种能够完美模拟人类的SSc,所以在选择合适的动物模型前应当明确自己的研究目的,广泛了解各种模型的特点,以便于选择最合适的研究模型,获得最有意义的结果。
(本文图2见封三)
[1]Kelley WN. Kelley’s textbook of rheumatology[M].Elsevier Health Sciences, 2012.
[2]李梦涛,侯勇,雷云霞,等. 合作与发展:系统性硬化症的未来--第一届国际系统性硬化症会议纪要[J]. 中华临床免疫和变态反应杂志, 2010, 4:151-154.
[3]Aso Y, Yoneda K, Kikkawa Y. Morphologic and biochemical study of pulmonary changes induced by bleomycin in mice[J].Lab Invest, 1976, 35:558-568.
[4]Yamamoto T, Takagawa S, Katayama I, et al. Animal model of sclerotic skin. I:Local injections of bleomycin induce sclerotic skin mimicking scleroderma[J].J Investigative Dermatol, 1999, 112:456- 462.
[5]Yamamoto T, Kuroda M, Nishioka K. Animal model of sclerotic skin. III:Histopathological comparison of bleomycin-induced scleroderma in various mice strains[J].Arch Dermatol Res, 2000, 292:535-541.
[6]Batteux F, Kavian N, Servettaz A. New insights on chemically induced animal models of systemic sclerosis[J].Curr Opin Rheumatol, 2011, 23:511-518.
[7]Yoshizaki A, Iwata Y, Komura K, et al. CD19 regulates skin and lung fibrosis via Toll-like receptor signaling in a model of bleomycin-induced scleroderma[J].Am J Pathol, 2008, 172:1650-1663.
[8]Kitaba S, Murota H, Terao M, et al. Blockade of interleukin-6 receptor alleviates disease in mouse model of scleroderma[J].Am J Pathol, 2012, 180:165-176.
[9]Okamoto Y, Hasegawa M, Matsushita T, et al. Potential roles of interleukin-17A in the development of skin fibrosis in mice[J].Arthritis Rheum, 2012, 64:3726-3735.
[10] Luzina IG, Kopach P, Lockatell V, et al. Interleukin-33 potentiates bleomycin-induced lung injury[J].Am J Respir Cell Mol Biol, 2013, 49:999-1008.
[11] Murota H, Hamasaki Y, Nakashima T, et al. Disruption of tumor necrosis factor receptor p55 impairs collagen turnover in experimentally induced sclerodermic skin fibroblasts[J].Arthritis Rheum, 2003, 48:1117-1125.
[12] Beyer C, Reichert H, Akan H, et al. Blockade of canonical Wnt signalling ameliorates experimental dermal fibrosis[J].Ann Rheum Dis, 2013, 72:1255-1258.
[13] Yoshizaki A, Yanaba K, Iwata Y, et al. Cell adhesion molecules regulate fibrotic process via Th1Th2Th17 cell balance in a bleomycin-induced scleroderma model[J].J Immunol, 2010, 185:2502-2515.
[14] Kokot A, Sindrilaru A, Schiller M, et al. α-melanocyte-stimulating hormone suppresses bleomycin-induced collagen synthesis and reduces tissue fibrosis in a mouse model of scleroderma:Melanocortin peptides as a novel treatment strategy for scleroderma?[J].Arthritis Rheum, 2009, 60:592-603.
[15] Wu M, Melichian DS, Chang E, et al. Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-γ[J].Am J Pathol, 2009, 174:519-533.
[16] Iwamoto N, Distler JHW, Distler O. Tyrosine kinase inhibitors in the treatment of systemic sclerosis:from animal models to clinical trials[J].Curr Rheumatol Rep, 2011, 13:21-27.
[17] Zhou CF, Yu JF, Zhang JX, et al. N-acetylcysteine attenuates subcutaneous administration of bleomycin-induced skin fibrosis and oxidative stress in a mouse model of scleroderma[J].Clin Exp Dermatol, 2013, 38:403- 409.
[18] Yoshizaki A, Yanaba K, Yoshizaki A, et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis[J].Arthritis Rheum, 2010, 62:2476-2487.
[19] Zhao L, Xiao K, Wang H, et al. Thalidomide has a therapeutic effect on interstitial lung fibrosis:evidence from in vitro and in vivo studies[J].Clin Exp Immunol, 2009, 157:310-315.
[20] Azhdari M, Baghaban-Eslaminejad M, Baharvand H, et al. Therapeutic potential of human-induced pluripotent stem cell-derived endothelial cells in a bleomycin-induced scleroderma mouse model[J].Stem Cell Res, 2013, 10:288-300.
[21] Hayashi S, Ikeda M, Kitamura Y, et al. UVA irradiation following treatment with topical 8-methoxypsoralen improves bleomycin-induced scleroderma in a mouse model, by reducing the collagen content and collagen gene expression levels in the skin[J].J Dermatol Sci, 2012, 67:20-25.
[22] Jaffee BD, Claman HN. Chronic graft-versus-host disease (GVHD) as a model for scleroderma:I. Description of model systems[J].Cell Immunol, 1983, 77:1-12.
[23] Ruzek MC, Jha S, Ledbetter S, et al. A modified model of graft-versus-host-induced systemic sclerosis (scleroderma) exhibits all major aspects of the human disease[J].Arthritis Rheum, 2004, 50:1319-1331.
[24] Servettaz A, Goulvestre C, Kavian N, et al. Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse[J].J Immunol, 2009, 182:5855-5864.
[25] Siracusa LD, McGrath R, Ma Q, et al. A tandem duplication within the fibrillin 1 gene is associated with the mouse tight skin mutation[J].Genome Res, 1996, 6:300-313.
[26] Denton CP, Lafyatis R. Overview of animal models[M]Scleroderma. Springer US, 2012:291-307.
[27] Christner PJ, Peters J, Hawkins D, et al. The tight skin 2 mouse[J].Arthritis Rheum, 1995, 38:1791-1798.
[28] Gershwin ME, Abplanal PH, Castles JJ, et al. Characterization of a spontaneous disease of white leghorn chickens resembling progressive systemic sclerosis (scleroderma)[J].J Exp Med, 1981, 153:1640-1659.
[29] Wick G, Andersson L, Hala K, et al. Avian models with spontaneous autoimmune diseases[J].Adv Immunol, 2006, 92:71-117.
[30] Eferl R, Hasselblatt P, Rath M, et al. Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2AP-1[J].Proc Natl Acad Sci UAS, 2008, 105:10525-10530.
[31] Maurer B, Busch N, Jüngel A, et al. Transcription factor fos-related antigen-2 induces progressive peripheral vasculopathy in mice closely resembling human systemic sclerosis[J].Circulation, 2009, 120:2367-2376.
[32] Bou-Gharios G, Garrett LA, Rossert J, et al. A potent far-upstream enhancer in the mouse pro alpha 2 (I) collagen gene regulates expression of reporter genes in transgenic mice[J].J Cell Biol, 1996, 134:1333-1344.
[33] Denton CP, Zheng B, Evans LA, et al. Fibroblast-specific expression of a kinase-deficient type II transforming growth factor β (TGFβ) receptor leads to paradoxical activation of TGFβ signaling pathways with fibrosis in transgenic mice[J].J Biol Chem, 2003, 278:25109-25119.
[34] Drab M, Verkade P, Elger M, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice[J].Science, 2001, 293:2449-2452.
[35] Del Galdo F, Lisanti MP, Jimenez SA. Caveolin-1, TGF-β receptor internalization, and the pathogenesis of systemic sclerosis[J].Curr Opin Rheumatol, 2008, 20:713.
