热休克蛋白70(hsp70)基因对利什曼原虫中国分离株的系统发育分析
2014-04-08张春莹黄玉霞胡孝素
张春莹,黄玉霞,袁 余,胡孝素,马 莹
利什曼病是一种由利什曼原虫引起的、经白蛉叮咬传播的寄生虫病。由于流行地域及生态环境复杂,人和其他宿主中存在多种利什曼原虫,其传播媒介和动物宿主呈多样性和交叉性。在我国,人的利什曼病按地理来源、传播媒介等流行特点可分为平原型、山丘型和荒漠型3种类型[1]。不同类型的利什曼病其传染源、病人临床表现等方面存在差异,预防控制措施也不同。由于不同种的利什曼原虫导致不同类型的利什曼病,因此快速、准确地对利什曼病病原体进行种株鉴定和系统发生研究对于利什曼病的诊治和预防非常重要。由于利什曼原虫的分型和种系发育复杂,而形态学方法难以区分出不同的种,因此更多的研究者倾向使用分子生物学方法对利什曼原虫进行鉴定、分型,但是不同研究者使用不同的分子生物学方法得到的结论不一致[2-4],因此中国利什曼原虫的种株鉴定仍处于不明确、有争议的状态。热休克蛋白70(heat shock protein 70,HSP70)是一种保守蛋白,该蛋白和其编码基因可用于不同利什曼原虫的种株鉴定和系统发育研究[5-6]。本研究通过对分离自我国各疫区不同宿主利什曼原虫的hsp70基因进行序列测定和分析,探讨利什曼原虫中国分离株种系发育关系,并将我国利什曼原虫和国外其他流行区的利什曼原虫hsp70基因序列共同分析,了解中国利什曼原虫与世界其他地区流行的利什曼原虫在种系发育和分子进化上的关系。
1 材料与方法
1.1材料
1.1.1利什曼原虫虫株来源 中国利什曼原虫分离株及WHO利什曼原虫参照株由本室保存,虫株详细信息及hsp70基因的GenBank登录号见表1。
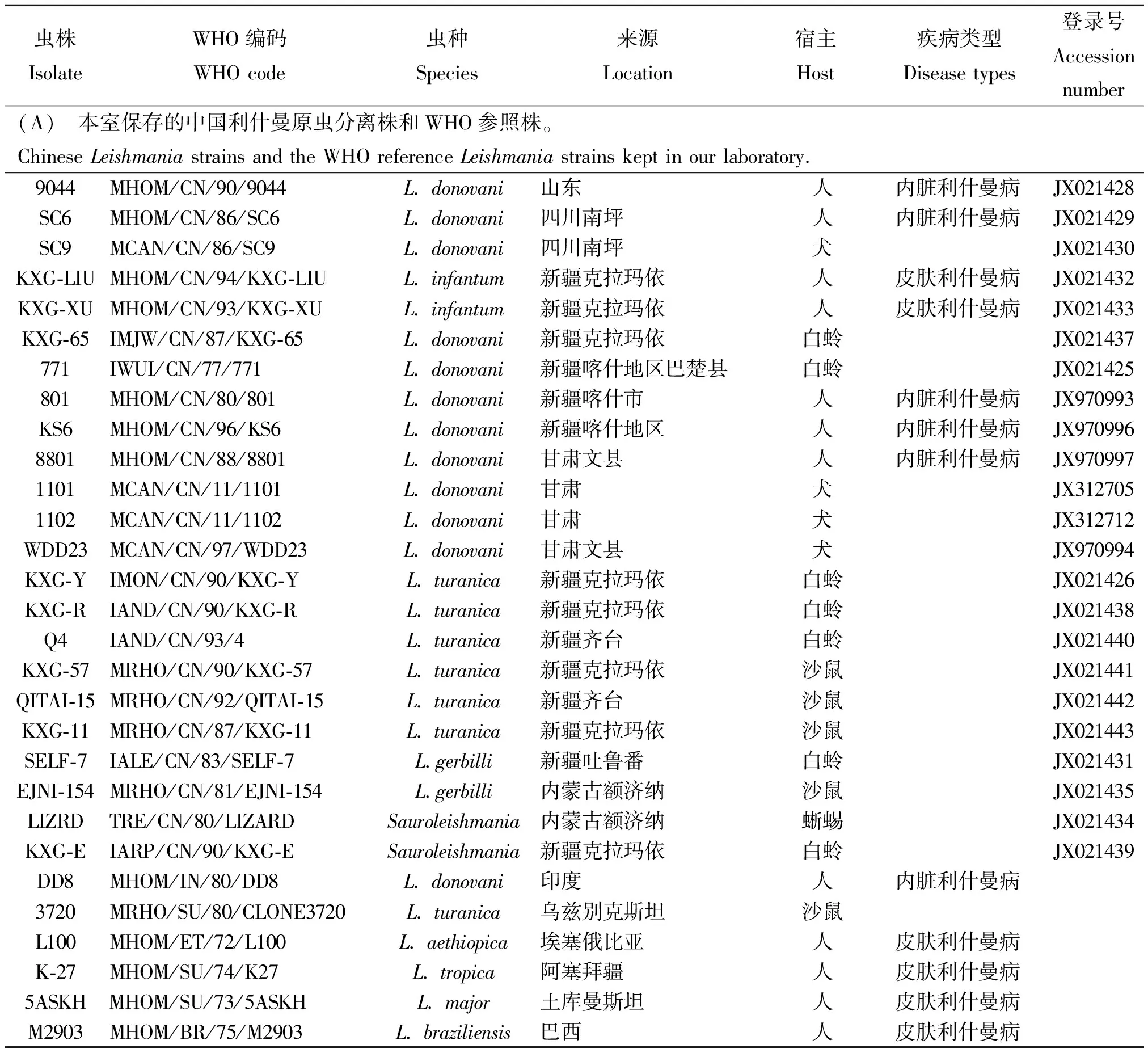
表1 利什曼原虫虫株详细信息及hsp70基因序列GenBank登录号
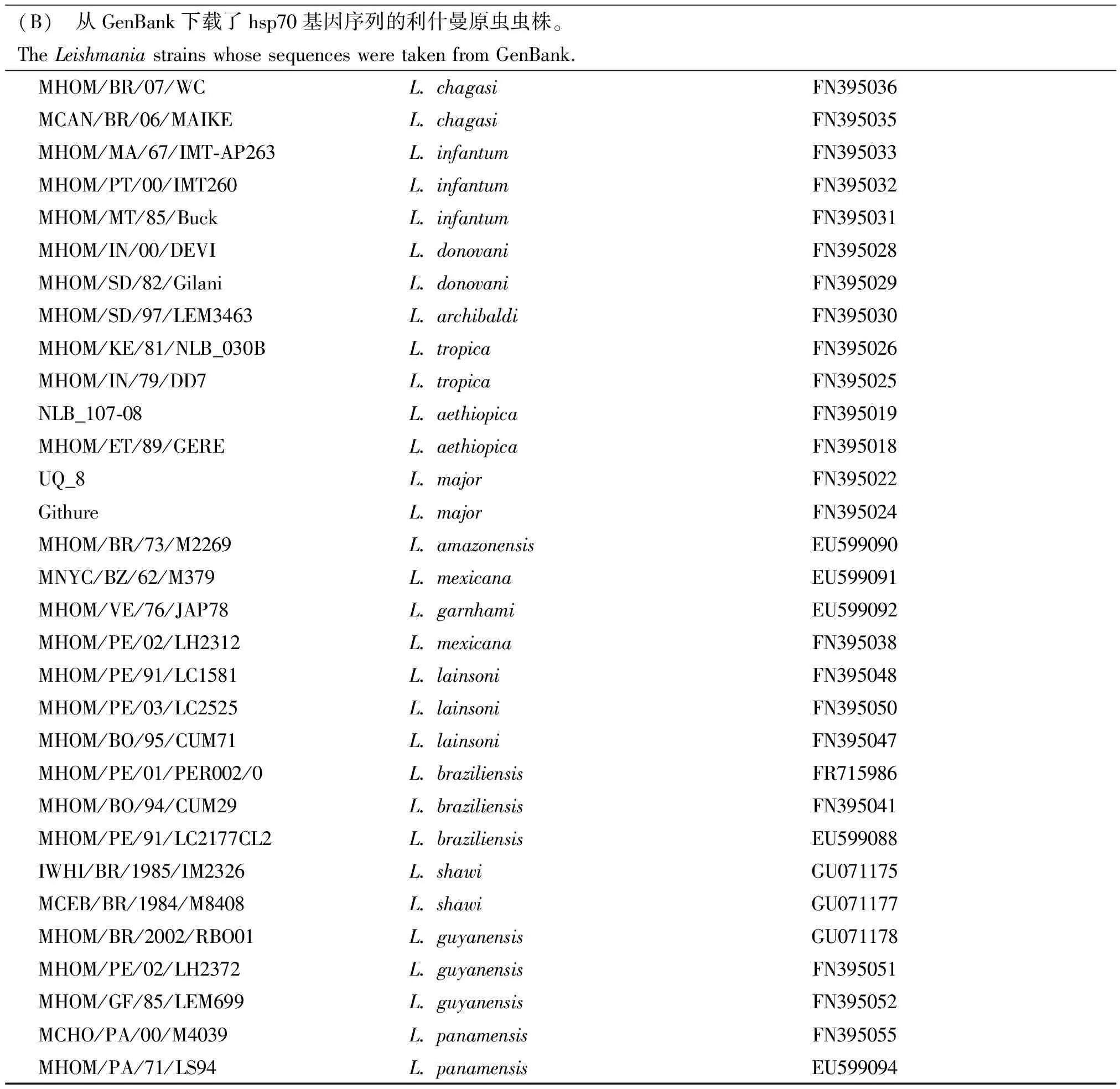
(B) 从GenBank下载了hsp70基因序列的利什曼原虫虫株。The Leishmania strains whose sequences were taken from GenBank.MHOM/BR/07/WCL. chagasiFN395036MCAN/BR/06/MAIKEL. chagasiFN395035MHOM/MA/67/IMT-AP263L. infantumFN395033MHOM/PT/00/IMT260L. infantumFN395032MHOM/MT/85/BuckL. infantumFN395031MHOM/IN/00/DEVIL. donovaniFN395028MHOM/SD/82/GilaniL. donovaniFN395029MHOM/SD/97/LEM3463L. archibaldiFN395030MHOM/KE/81/NLB_030BL. tropicaFN395026MHOM/IN/79/DD7L. tropicaFN395025NLB_107-08L. aethiopicaFN395019MHOM/ET/89/GEREL. aethiopicaFN395018UQ_8L. majorFN395022GithureL. majorFN395024MHOM/BR/73/M2269L. amazonensisEU599090MNYC/BZ/62/M379L. mexicanaEU599091MHOM/VE/76/JAP78L. garnhamiEU599092MHOM/PE/02/LH2312L. mexicanaFN395038MHOM/PE/91/LC1581L. lainsoniFN395048MHOM/PE/03/LC2525L. lainsoniFN395050MHOM/BO/95/CUM71L. lainsoniFN395047MHOM/PE/01/PER002/0L. braziliensisFR715986MHOM/BO/94/CUM29L. braziliensisFN395041MHOM/PE/91/LC2177CL2L. braziliensisEU599088IWHI/BR/1985/IM2326L. shawiGU071175MCEB/BR/1984/M8408L. shawiGU071177MHOM/BR/2002/RBO01L. guyanensisGU071178MHOM/PE/02/LH2372L. guyanensisFN395051MHOM/GF/85/LEM699L. guyanensisFN395052MCHO/PA/00/M4039L. panamensisFN395055MHOM/PA/71/LS94L. panamensisEU599094
1.2方法
1.2.1利什曼原虫基因组DNA(nDNA)的提取 复苏液氮保存的利什曼原虫虫株转入NNN培养基,扩大培养后用饱和酚-氯仿法提取基因组DNA[7],-20℃保存备用。
1.2.2hsp70基因PCR扩增和序列测定 PCR引物为:正向 3′-GACGGTGCCTGCCTACTTCAA-5′,反向3′- CCGCCCATGCTCTGGTACATC-5′。引物由英潍捷基(invitrogen,上海)贸易有限公司合成。PCR扩增反应体系为25 μL,扩增条件为:95 ℃预变性5 min,95 ℃变性60 s,Y℃退火60 s,72 ℃延伸90 s,共35个循环,最后72 ℃延伸10 min。取3 μL PCR产物经2%琼脂糖凝胶(含5 μL/100 mL溴化乙锭)电泳后,用紫外凝胶成像仪观察扩增条带。将预期大小的PCR产物进行序列测定,测序工作由华大基因完成。
1.2.3种系发育分析:采用ContigExpress软件(http://www.contigexpress.com/)进行双向序列拼接,根据峰形图对序列信息进行手工校正。使用分子进化遗传分析工具(molecular evolution genetics analysis, MEGA)[8]进行多序列比对, 将整理后的序列提交GenBank以获得序列登录号。采用jModelTest v.0.1.1软件[9]分析核苷酸序列,选择合适的进化模型,用于构建系统发育树。用MEGA 5.0软件构建Neighbor-Joining (NJ)树,推测中国利什曼原虫分离株间的系统发育情况,采用自展分析法(bootstrapping analysis)对构建的系统发育树进行评估。从在线基因数据库(NCBI、EMBL或DDBJ)中下载其他利什曼原虫分离株相应编码基因的核苷酸序列资料,将本研究获得的序列和下载的序列一起比对,重建系统发生树,推测中国分离株在世界利什曼原虫分离株的系统发生和分子进化中的地位。
2 结 果
2.1PCR扩增产物 经2%的琼脂糖凝胶电泳,各利什曼原虫分离株均扩增出与预期长度一致的hsp70基因的清晰条带,无非特异性条带出现,空白对照为阴性。
2.2hsp70基因序列测定及同源性比较 用双脱氧核糖核酸末端终止法进行双向测序,根据峰形图对序列信息进行校正,当峰形与碱基不一致时,通过人工比对进行手工校正。采用ContigExpress软件进行双向序列拼接,获得约1 300 bp长度的hsp70基因序列。将整理后的hsp70基因序列提交GenBank,获得序列登录号见表1。使用MEGA 5.0软件对hsp70基因序列进行多序列比对,WHO参照株巴西利什曼原虫(L.braziliensis)M2903表现出最大的多态性。在所分析的1 278 bp长度的hsp70基因序列中,有95个多态性位点,当排除M2903后比较,出现56个多态性位点。Z-test检测压力选择,hsp70基因在进化中保持中性,不受选择压力的影响。
2.3种系发育分析 MEGA 5.0 软件构建邻接树(Neighbor-Joining tree),树形结构显示中国各疫区不同宿主的利什曼原虫分离株聚集为不同种类的4群(图1)。13株人来源或犬来源的利什曼原虫与WHO参照株DD8聚为A群,为杜氏利什曼原虫(L.donovani)。来自新疆沙鼠或白蛉的分离株与WHO参照株3720聚为B群,为都兰利什曼原虫(L.turanica);分离自新疆白蛉的利什曼原虫(SELF-7)和分离株内蒙古沙鼠的利什曼原虫(EJNI-154)聚为C群,为沙鼠利什曼原虫(L.gerbilli),在系统发育树中C群与B群相距较近;D群中包含两株,分别为一株内蒙古蜥蜴的分离株LIZRD和一株新疆白蛉的分离株KXG-E,为蜥蜴利什曼原虫(Sauroleishmania)。D群与A、B、C群的虫株差异较大,因而在系统发育树中单独聚为较远的一支。WHO参照株K-27(热带利什曼原虫,L.tropica)、5AS(硕大利什曼原虫,L.major)和L100(埃塞俄比亚利什曼原虫,L.eathiopica)由于与中国分离株存在较大差异而归入其他分支,M2903(巴西利什曼原虫,L.braziliensis)是新世界分离株,为L.Viannia。
联合分析国内外60株利什曼原虫的hsp70序列(见图2),结合Frage等5]和Schönian等[10]对利什曼原虫分型的观点,中国分离株聚集为L. (Leishmania)亚属的L.donovani和L.major两个种,其中A群为L.donovani,根据hsp70基因碱基差异,该群又分为A1、A2两个亚群,A1亚群包含6株中国分离株和3株其他国家的杜氏利什曼原虫指名亚种,A2亚群包含7株中国分离株和5株其他国家的杜氏利什曼原虫婴儿亚种; 我国的B群(L.turanica)和C群(L.gerbilli)为L.major,我国的D群为利什曼原虫的另外一个亚属L. (Sauroleishmania)。
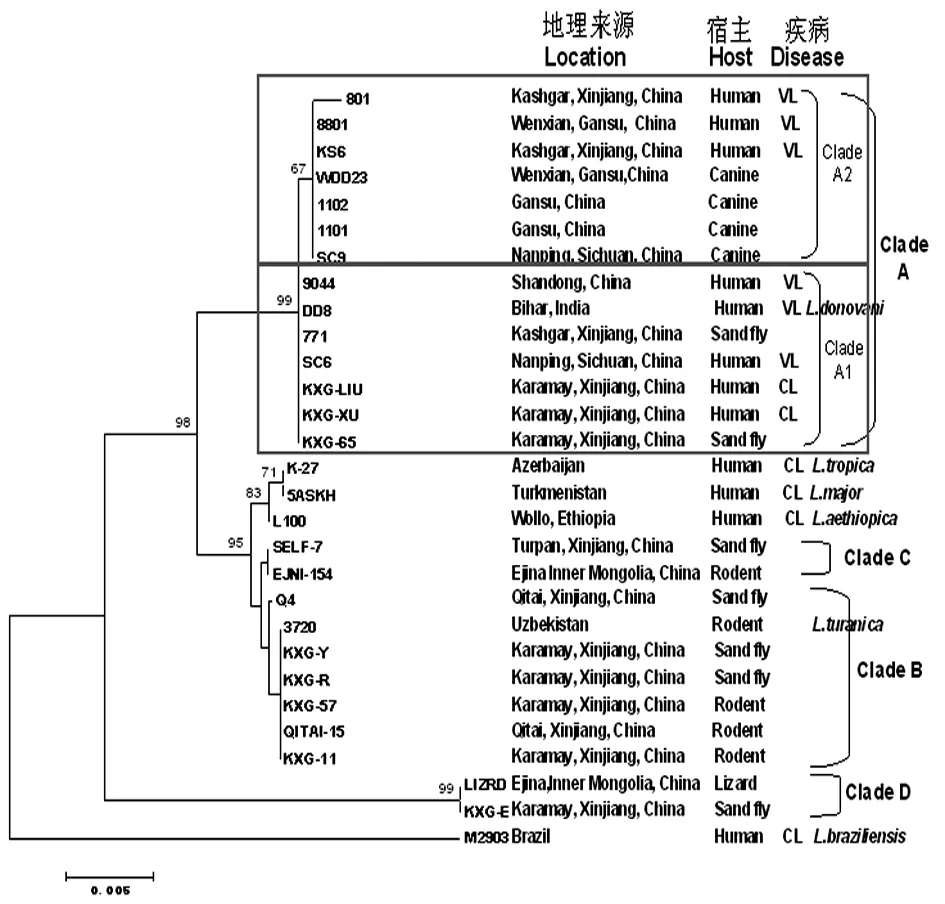
图129株利什曼原虫的hsp70基因序列构建的Neighbor-Joining无根树
Fig.1Neighbor-joiningunrootedtreeconstructedbasedonthesequencesofhsp70genefor29Leishmaniaisolatesinthisstudy(23ChineseLeishmaniastrainsand6WHOreferencestrains)
The Kimura-2-parameter model in MEGA 5.0 was used. Numbers above branches correspond to bootstrap values based on 1 000 replicates. The strains were designated by their names.
3 讨 论
利什曼病(leishmaniasis)对人类健康危害严重。在我国,本病曾流行于17个省、市、自治区,目前,平原疫区的山东和江苏等地的利什曼原虫已消除,但新疆喀什绿洲的利什曼病仍处于活跃状态[11];山丘疫区和荒漠疫区的利什曼病流行较严重,不断有新感染病例出现[12]。我国不同流行疫区的利什曼病病原体有差异,有学者[13-14]以往根据黑热病流行的地貌类型特点和传染源种类将我国利什曼病流行区分为:平原型(人源型)、山丘型(犬源型)和荒漠型(野生动物源型),并且分别认为平原型的虫种为杜氏利什曼原虫,山丘型和荒漠型的病原为婴儿利什曼原虫,但近年来国内外许多研究认为中国的利什曼原虫种类更加复杂多样[15]。热休克蛋白70(hsp70)基因已广泛应用于多种寄生虫的种系发育研究,因此本研究对利什曼原虫分离株的hsp70基因进行分析,研究我国利什曼原虫分离株之间以及我国利什曼原虫与世界其他流行区的利什曼原虫之间的种系发育情况和进化关系。

图229株利什曼原虫与GenBank下载的31株利什曼原虫的lack基因序列构建的Neighbor-Joining无根树。
Fig.2Neighbor-joiningtreeconstructedbasedonthesequencesof29Leishmaniaisolates(23Chineseisolatesand6WHOreferenceisolates)inthisstudyand31isolateswhosesequencesweretakenfromGenBank
The Kimura-2-parameter model in MEGA 5.0 software was used. Bootstrap values based on 1 000 replicates are shown above in each branch. The strains were designated by their names for those in this study and by the accession numbers for those in other studies.
基于hsp70基因构建的系统发育树显示,中国各疫区利什曼原虫分离株聚集为4群 (见图1),分别为A群杜氏利什曼原虫 (L.donovani),B群都兰利什曼原虫(L.turanica),C群沙鼠利什曼原虫(L.gerbilli)和D群蜥蜴利什曼原虫(Sauroleishmania),其中分离自人的虫株均归为杜氏利什曼原虫。有研究指出[16],宿主来源不同影响利什曼原虫虫株的系统发育,即利什曼病的发生与宿主有关,如在中国发现的L.turanica(B群)仅来自于沙鼠和白蛉,在国内尚未见感染人的L.turanica。而从蜥蜴体内分离的病原体为蜥蜴利什曼原虫(D群),这种虫种跟宿主相关的流行病学特点可能受许多复杂因素的影响,目前这些因素尚未一一阐明。联合比对分析本研究29株利什曼原虫hsp70序列和基因数据库中下载的31株世界其他流行区利什曼原虫的hsp70序列,重建系统发育树,见图2。利什曼原虫属有三个亚属:L. (Leishmania),L. (Sauroleishmania)和L. (Viannia)。中国分离株有两株为L. (Sauroleishmania)亚属,其余虫株均为L. (Leishmania)亚属。在杜氏利什曼原虫(L.donovani)中,hsp70基因将中国利什曼原虫分为两个亚支:杜氏利什曼原虫指名亚种(L.donovani)和杜氏利什曼原虫婴儿亚种(L.infantum)。分离自四川犬的原虫(SC9)、甘肃人和犬的原虫(8801、1101、1102)及分离自新疆喀什地区人的利什曼原虫(801、KS6)为杜氏利什曼原虫婴儿亚种(L.infantum),分离自四川人的原虫(SC6)、山东人的原虫(9044)及新疆人(皮肤利什曼病)和白蛉的利什曼原虫(KXG-LIU、KXG-XU、771、KXG-65)为杜氏利什曼原虫指名亚种(L.donovani)。新疆克拉玛依的沙鼠和白蛉体内分离出的虫株为都兰利什曼原虫(L.turanica),与国内其他研究方法得出的结论一致[15]。都兰利什曼原虫和沙鼠利什曼原虫的保虫宿主为沙鼠,传播媒介为白蛉,两种原虫的关系相近,因此在系统发育树中,B群和C群的分支邻近。
虫株SC6和SC9分别分离自四川南坪的人和犬,二者均聚为L.donovani复合体,但仍因存在一定的差异而归入两个亚群(A1和A2),在利什曼原虫感染人和犬时,可能由于虫株对人、犬的侵入和免疫逃避反应不同,为适应宿主而达到虫体在宿主体内感染的平衡状态,病原体会发生一定的变异,从而导致SC6、SC9出现个别核苷酸序列的差异。但尚不清楚这种差异是否是一种稳定的现象存在?需要更多来自四川疫区人和犬的利什曼原虫分离株来证实。而Sun K等[17]通过7SL RNA序列得到的结论认为分离自四川犬和人的利什曼原虫为利什曼属的未定种,该未定种的利什曼原虫在系统发育树中与蜥蜴利什曼原虫最相近,与本研究的分型结果不同。本研究中仅有两株四川的利什曼原虫分离株(SC6、SC9),由于不同的基因其侧重的研究靶点不同,基因所受到的选择压力或基因漂移的作用不同,对利什曼原虫的进化史推测到的结果可能会有分歧,不能排除研究中基因选择偏倚对分子分型的影响,因此需要研究更多来自四川疫区人和犬的利什曼原虫分离株,联合扩增其多个保守基因,来证实分离自四川的利什曼原虫的种属情况。
本研究结果与以往国内学者的结论有差异,以往研究学者是根据利什曼病流行区的地理景观、流行特征和传染源种类等特点将我国的利什曼原虫进行区分,而本文从分子水平上对虫株进行分类,分辨率高且快速简便,有利于准确查找传染源头,追踪病原体的进化过程,控制疾病的传染。
参考文献:
[1]Guan LR. Present situation of visceral leishmaniasis and prospect for its control in China[J]. Chin J Parasitol Parasit Dis, 2009, 27(5): 394-397. (in Chinese)
管立人. 我国内脏利什曼病的现状和对防治工作的展望[J]. 中国寄生虫学与寄生虫病杂志, 2009, 27(5):394-397.
[2]Wang JY, Gao CH, Yang YT, et al. Evaluation of a repetitive DNA sequence ofLeishmaniaon the identification ofLeishmaniaisolates in China[J]. Chin J Zoonoses, 2005, 21(4): 304-308. (in Chinese)
汪俊云, 高春花, 杨玥涛, 等. 利什曼原虫-DNA重复序列在虫种鉴定上的价值分析[J]. 中国人兽共患病杂志, 2005, 21(4):304-308.
[3]Zhang T, Hu XS, Jing BQ, et al. Sequence analysis of SSUrDNA variable region fo cutaneous Leishmaniasis pathogen from Xinjiang, China[J]. Chin J Parasit Dis Ctrl, 1998, 11(4): 279-283. (in Chinese)
章涛, 胡孝素, 敬保迁, 等. 我国新疆皮肤利什曼病病原体SSU rDNA多变区序列分析[J]. 中国寄生虫病防治杂志, 1998, 11(4):279-283.
[4]Chen JP, Hu XS, Zheng XL, et al. Study on the molecular hybridization in kDNA of 3Leishmaniaspecies and the pathogenic agent of Cutaneous leishmaniasis in Xinjiang, China[J]. Chin J Vect Biol Ctrl, 1997, 8(1): 54-57. (in Chinese)
陈建平, 胡孝素, 郑学礼, 等. 三种利什曼原虫和我国新疆皮肤利什曼原虫kDNA分子杂交实验研究[J]. 中国媒介生物学及控制杂志, 1997, 8(1):54-57.
[5]Fraga J, Montalvo AM, De Doncker S, et al. Phylogeny ofLeishmaniaspecies based on the heat-shock protein 70 gene[J]. Infect Genet Evol, 2010, 10(2): 238-245. DOI: 10.1016/j.meegid.2009.11.007
[6]Montalvo AM, Fraga J, Monzote L, et al. Heat-shock protein 70 PCR-RFLP: a universal simple tool forLeishmaniaspecies discrimination in the New and Old World[J]. Parasitology, 2010, 137(8): 1159-1168. DOI: 10.1017/S0031182010000089
[7]Schonian G, Schweynoch C, Zlateva K, et al. Identification and determination of the relationships of species and strains within the genusLeishmaniausing single primers in the polymerase chain reaction[J]. Mol Biochem Parasitol, 1996, 77(1): 19-29.
[8]Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods[J]. Mol Biol Evol, 2011, 28(10): 2731-2739. DOI: 10.1093/molbev/msr121
[9]Posada D. jModelTest: phylogenetic model averaging[J]. Mol Biol Evol, 2008, 25(7): 1253-1256. DOI: 10.1093/molbev/msn083
[10]Schonian G, Mauricio I, Cupolillo E. Is it time to revise the nomenclature ofLeishmania?[J]. Trends Parasitol, 2010, 26(10): 466-469. DOI: 10.1016/j.pt.2010.06.013
[11]Tian T, Wu WP, Wang LY, et al. Epidemiological analysis on visceral leishmaniasis in China during 2008-2011[J]. Int J Med Parasit Dis, 2012, 39(4): 223-226. (in Chinese)
田添, 伍卫平, 王立英, 等. 2008-2011年我国内脏利什曼病流行病学分析[J]. 国际医学寄生虫病杂志, 2012, 39(4):223-226.
[12]Wang JY, Gao CH, Yang YT, et al. An outbreak of the desert sub-type of zoonotic visceral leishmaniasis in Jiashi, Xinjiang Uygur Autonomous Region, People's Republic of China[J]. Parasitol Int, 2010, 59(3): 331-337. DOI: 10.1016/j.parint.2010.04.002
[13]Lv HG, Zhong L, Guan LR, et al. Separation of ChineseLeishmaniaisolates into five genotypes by kinetoplast and chromosomal DNA heterogeneity[J]. Am J Trop Med Hyg, 1994, 50(6): 763-770.
[14]Wang JY, Qu JQ, Wang RQ. Analysis on homology in several isolates ofLeishmaniafrom Karamay, Xinjiang[J]. Chin J Parasitol Parasit Dis, 1996, 14(4): 266-269. (in Chinese)
汪俊云, 瞿靖琦, Rongqi Wang, 等. 新疆克拉玛依地区几株利什曼原虫分离物的同源性分析[J].中国寄生虫学与寄生虫病杂志, 1996, 14(4):266-269.
[15]Yang BB, Guo XG, Hu XS, et al. Species discrimination and phylogenetic inference of 17 ChineseLeishmaniaisolates based on internal transcribed spacer 1 (ITS1) sequences[J]. Parasitol Res, 2010, 107(5): 1049-1065. DOI: 10.1007/s00436-010-1969-9
[16]Dujardin JC. Risk factors in the spread of leishmaniases: towards integrated monitoring?[J]. Trends Parasitol, 2006, 22(1): 4-6.
[17]Sun K, Guan W, Zhang JG, et al. Prevalence of canine leishmaniasis in Beichuan County, Sichuan, China and phylogenetic evidence for an undescribedLeishmaniasp. in China based on 7SL RNA[J]. Parasit Vectors, 2012, 5(1): 75. DOI: 10.1186/1756-3305-5-75
