ERK1/2在缺血再灌注损伤肺细胞凋亡中的作用及缺血后处理的干预
2014-03-23陈海娥马迎春黄林静何金波宋冬郑梦晓应磊王万铁
陈海娥,马迎春,黄林静,何金波,宋冬,郑梦晓,应磊,王万铁
(温州医科大学 缺血/再灌注损伤研究所、病理生理学教研室,浙江 温州 325035)
·论 著·
ERK1/2在缺血再灌注损伤肺细胞凋亡中的作用及缺血后处理的干预
陈海娥,马迎春,黄林静,何金波,宋冬,郑梦晓,应磊,王万铁
(温州医科大学 缺血/再灌注损伤研究所、病理生理学教研室,浙江 温州 325035)
目的:探究细胞外信号调节激酶1/2(ERK1/2)在缺血/再灌注(IR)损伤肺细胞凋亡中的作用及缺血后处理的干预。方法:雄性SD大鼠,随机分成5组(n=8),即对照组(C组)、肺缺血/再灌注组(IR组)、肺缺血/再灌注+缺血后处理组(IPO组)、缺血后处理+溶剂对照组(D组)、缺血后处理+U0126组(U组)。分别于再灌注2 h留取左肺组织,电镜观察肺组织超微结构改变;原位末端标记(TUNEL)法检测肺细胞凋亡情况并计算凋亡指数(AI);RT-PCR法、免疫组化法测定肺组织Bax、Bcl-2基因和蛋白的表达。结果:与C组相比,IR组肺组织AI、Bax基因及蛋白表达均显著升高(P<0.05),电镜下肺细胞均发生明显损伤;Bcl-2、Bcl-2/Bax基因及蛋白表达显著降低(P<0.05);IPO组、D组、U组与IR组相比,肺组织AI、Bax基因及蛋白表达均显著降低(P<0.05),电镜下肺细胞损伤情况有所改善;Bcl-2、Bcl-2/Bax显著升高;D组与IPO组比较各项指标差异均无统计学意义(均P>0.05),U组与IPO组相比,肺组织AI值显著升高(P<0.05),电镜下肺上皮细胞超微结构破坏较严重,胞内细胞器不完整;Bcl-2基因及蛋白表达明显降低,Bax基因及蛋白表达明显升高,Bcl-2/Bax显著降低。结论:IR抑制了MAPK家族中的ERK1/2激活,导致大鼠肺组织结构严重破坏,细胞大量凋亡;IPO可以通过激活ERK1/2通路,改善其结构破坏和细胞凋亡。
缺血/再灌注;缺血后处理;肺;细胞凋亡;细胞外信号调节激酶1/2;U0126;Bcl-2;Bax;大鼠
随着移植技术的广泛开展,缺血/再灌注(ischemia reperfusion,IR)损伤成为了阻止移植成功的主要难题,肺缺血/再灌注是影响心肺移植成功与否的重大问题,其发生机制非常复杂,目前认为主要与氧自由基损伤、细胞凋亡、细胞内钙超载损伤及内皮细胞的激活、炎性细胞的聚集激活、细胞黏附分子的释放等因素有关[1]。缺血后处理(ischemic postconditioning,IPO)是在再灌注早期进行的一系列快速的血流中断/再通的过程,其可以改变再灌注血流动力学并对缺血再灌注器官产生保护作用,是近年发现的一种重要内源性保护机制。已有研究表明IPO对肺IR有明显的保护作用,但其机制仍不清楚[2]。
细胞外信号调节激酶1/2(extracellular signal-regulated kinase 1/2,ERK1/2)是丝裂原活化蛋白激酶(mitogen-activatd protein kinase,MAPK)家族的一员,其基本的信号传递步骤是:Ras-Raf-MEK-ERK。ERK通路在细胞增殖、分化、抗凋亡中发挥重要作用,可被多种刺激(如神经递质、氧化应激、缺血等)激活[3]。有研究表明ERK1/2在心脏IPO中被激活而发挥保护作用[4],尚未有研究表明其是否在肺IPO中发挥作用,本实验意在探明ERK1/ 2在肺IPO中的作用。
1 材料和方法
1.1 实验动物SPF级SD大鼠40只,雄性,体质量200~250 g,由上海斯莱克动物实验中心提供[实验动物使用许可证号:SCXK(沪)2012-0002]。动物实验方案经温州医科大学实验动物伦理委员会审核并同意实施。
1.2 主要试剂U0126(货号U120,美国Sigma公司),原位细胞凋亡检测试剂盒(LOT:13033000,德国Roche公司),Trizol提取液(美国Life technologies公司),反转录试剂盒(美国Fermentas Life Science公司),PCR试剂盒(美国Fermentas Life Science公司),Bcl-2鼠多克隆抗体(美国santa cruz公司),Bax鼠多克隆抗体(美国santa cruz公司),即用型SABC过氧化物酶试剂盒(武汉博士德公司),DAB显色试剂盒(北京中杉金桥生物科技有限公司),脱氧核糖核酸酶I(美国Sigma公司),其余均为市售分析纯。
1.3 动物模型复制按照已发表文献[2]报道的方法复制大鼠原位肺IR和IPO动物模型。大鼠称重后,按8 mL/kg体质量20%乌拉坦腹腔注射麻醉。麻醉后气管插管,连接小动物呼吸机,行机械通气,呼吸机参数设置为:潮气量7~8 mL/kg,呼吸频率70 b/min,吸呼比3:2。消毒左胸部皮肤,分离筋膜和肌肉,断开第3、第4根肋骨,打开胸腔,暴露左肺,游离左肺门,动脉夹夹闭30 min,即为缺血期,松开动脉夹,即为再灌注期,再灌注时间为2 h,如此即成功复制在体原位单肺IR模型;再灌注前给予3个循环的缺血30 s/再灌注30 s处理,结束后再灌注2 h,即为IPO组。C组游离左肺门后不夹闭肺门,观察2.5 h。
1.4 实验分组随机将实验动物分为5组(每组8只):对照组(C组),IR组,IPO组,IPO+0.4%二甲基亚砜(DMSO)的PBS溶液对照组(D组),IPO+U0126组(U组)。U0126用药剂量为0.3 mg/kg体质量,将0.3 mg U0126溶于7.5 mL 0.4% DMSO的PBS溶液中,使其充分溶解,缺血前尾静脉注入大鼠体内[5],余处理同IPO组;D组于缺血前尾静脉注入7.5 mL/ kg 0.4% DMSO的PBS溶液,余处理同IPO组。各组实验结束后留取左肺组织标本。
1.5 肺组织电镜检测依据文献[6]所述方法进行,即各组实验结束后,处死动物,取左肺近肺门处约1 mm×1 mm×1 mm大小的肺组织若干小块,立即经2.5%戊二醛前固定,再依次经1%锇酸后固定,1%醋酸铀块染,酒精梯度脱水,丙酮浸透,Epon 812包埋聚合,半薄切片和超薄切片,经醋酸铀和硝酸铅双重染色后,于电镜下观察各标本超微结构改变。
1.6 原位末端标记(TUNEL)法检测依据文献[7] Roche公司提供的试剂盒说明书所述方法进行操作。凋亡细胞胞核经DAB染色呈棕褐色,未凋亡细胞胞核复染后呈蓝紫色。计数5个高倍镜(×400)下的凋亡细胞数。每张片子至少观察500个细胞,计算每100个细胞内的阳性细胞,即凋亡指数(apoptotic index,AI)。
1.7 RT-PCR法测定肺组织Bax、Bcl-2基因的表达Trizol法提取肺组织总RNA。测定总RNA浓度。按照反转录试剂盒说明书反转录合成cDNA。再按照PCR试剂盒说明书进行cDNA扩增。内参及各目的基因的序列见表1。PCR反应条件为:起始变性,94 ℃,3 min;变性,94 ℃,30 s;退火,55 ℃(β-actin和Bax)/61.6 ℃(Bcl-2),30 s;延伸,72 ℃,1 min;终止延伸,72 ℃,5 min。循环数β-actin和Bax各30次,Bcl-2 33次。各目的基因扩增产物片段长度为:β-actin:378 bp;Bax:337 bp;Bcl-2:411 bp。用各基因扩增产物的密度与β-actin基因扩增产物的密度比值(即Bcl-2 mRNA/β-actin mRNA、Bax mRNA/β-actin mRNA)表示各基因表达水平。

表1 引物序列
1.8 免疫组化法检测标本中Bax、Bcl-2蛋白的表达水平及其定位按照试剂盒所提供的说明书和文献[8]中所述步骤进行操作。标本上呈棕褐色显色的部分为Bcl-2、Bax蛋白阳性表达。采用美国IPP6.0(Image-pro plus)彩色图像分析系统测定阳性部位及背景的光密度值,以阳性部位的光密度值减背景的光密度值代表阳性部位的吸光度(optical density,OD)值。
1.9 统计学处理方法采用SPSS17.0软件。计量资料均进行正态性检验,以±s表示。多组样本均数比较进行方差齐性检验,组间比较采用单因素方差分析,方差齐性者两两比较采用LSD法,方差不齐者进行Dunnet’s t检验。双变量相关性分析,采用Bivariate过程的Pearson相关分析法。P<0.05为差异有统计学意义。
2 结果
2.1 电镜下肺组织超微结构的变化C组电镜下细胞结构完整,细胞器清晰可见,核内染色质均匀,未见核固缩碎裂或溶解;IR组超微结构损伤较严重,可见核碎裂、溶解,细胞器结构破坏严重,肺泡I I型上皮细胞内线粒体肿胀、脊消失,板层小体空泡化;IPO组和D组损伤有所减轻,未见明显核碎裂、溶解,细胞间连接紧密,肺泡I I型上皮细胞内板层小体数量尚可,未见明显空泡化,细胞器结构无破坏;U组细胞结构略有破坏,细胞间连接较疏松,部分细胞核内出现碎裂、溶解,核染色质边集,肺泡I I型上皮细胞微绒毛略有脱落,线粒体肿胀(见图1)。
2.2 各组细胞凋亡情况及AI值的变化与C组(11.66 ±1.11)相比,IR组AI值(35.65±0.76)显著升高,D组(25.74±0.94)、U组(30.86± 1.13)、IPO组(26.26±0.95)AI值显著低于IR组(均P<0.05),D组与IPO组相比无明显差异(P>0.05),U组显著高于IPO组(P<0.05)。凋亡细胞主要为肺泡上皮细胞与血管内皮细胞(见图2)。
2.3 肺组织Bax、Bcl-2基因表达水平的变化在C组Bcl-2呈高水平表达、Bax表达不明显,IR组较C组Bcl-2表达明显下降、Bax明显上调,同时Bcl-2/Bax的比值降低(均P<0.05);与IR组比较,D组、U组、IPO组Bcl-2 mRNA表达增强,Bax mRNA表达减弱,Bcl-2/Bax的比值增高(均P<0.05),D组与IPO组相比差异无统计学意义(P>0.05);U组与IPO组比较,Bcl-2表达下降、Bax表达上调,二者比值下降(见图3)。

图2 TUNEL法示各组肺组织细胞凋亡情况(×400)
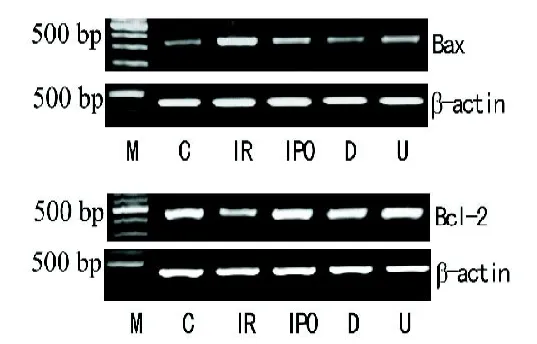
图3 ART-PCR法示Bcl-2 mRNA、Bax mRNA的表达

图3 B柱状图示Bcl-2 mRNA、Bax mRNA表达及Bcl-2/Bax比值变化
2.4 肺组织 Bax、Bcl-2蛋白的表达水平的变化及定位在C组Bcl-2蛋白呈高水平表达,Bax蛋白表达不明显,IR组表达较C组Bcl-2表达明显下降,Bax明显上调,同时Bcl-2/Bax的比值降低(均P<0.05);与IR组比较,D组、U组、IPO组Bcl-2蛋白表达增强,Bax表达减弱,Bcl-2/Bax的比值增高(P<0.05),D组与IPO组相比差异无统计学意义(P>0.05),U组Bcl-2表达较IPO组明显降低、Bax表达明显升高,二者比值降低。Bcl-2与Bax阳性细胞主要分布于血管内皮细胞、肺泡上皮细胞及支气管上皮细胞(见图4-6)。
2.5 相关性分析肺组织AI与Bax基因及蛋白表达之间呈正相关(r分别为0.796,0.895,均P<0.01,n=40);肺组织AI与肺组织Bcl-2基因及蛋白、Bcl-2/Bax基因及蛋白呈负相关(r分别为-0.748,-0.797,-0.897,-0.960,均P<0.01,n=40)。
3 讨论
细胞凋亡又被称为程序化细胞死亡,是细胞接受某种信号后或受到某些因素剌激后的一种主动的,由一些凋亡相关基因相互作用的细胞死亡过程。近年来,国内外学者的大量研究发现,细胞凋亡参与了肺IR损伤[1]。本实验中,IR组AI值较C组明显升高,肺组织超微结构损伤较C组严重,这些充分证明了以上结论。
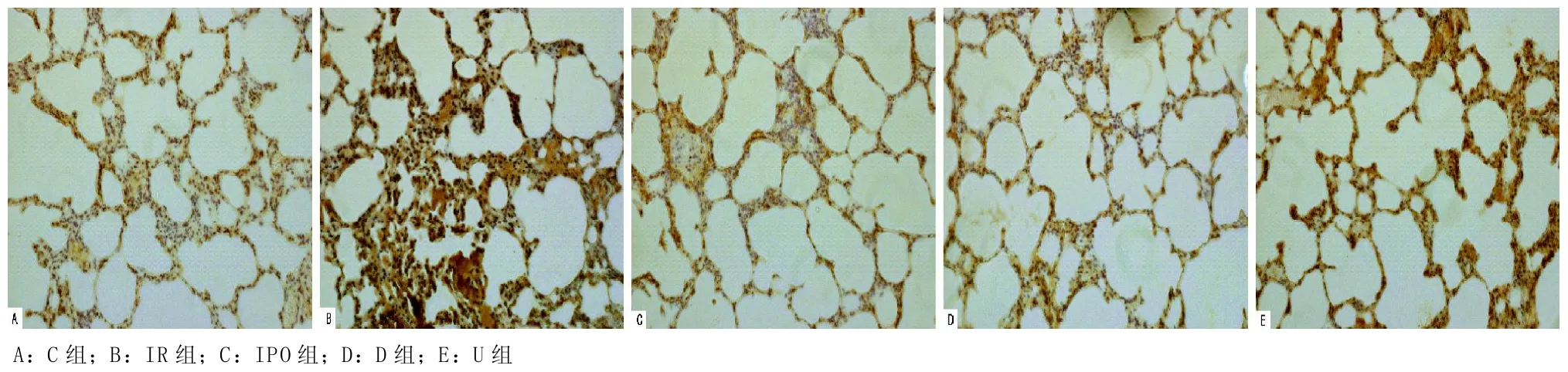
图4 各组肺组织Bax蛋白表达情况(免疫组化法,×200)

图5 各组肺组织Bcl-2蛋白表达情况(免疫组化法,×200)

图6 IPP6.0计算各组肺组织Bcl-2、Bax蛋白表达情况及Bcl-2/Bax比值
Ng等[1]报道,触发细胞凋亡的途径主要有死亡受体途径(外源性途径)和线粒体-细胞色素C(cytochrome C,Cyt c)途径(内源性途径)。其中外源性途径通过特定的受体-配体相互作用,比如Fas/Fas-L、血管紧张素I I和肿瘤坏死因子(TNF)/受体,导致一系列的细胞内凋亡级联激活主要是半胱氨酸天冬氨酸蛋白酶(caspases);内源性途径主要是通过Bcl-2家族基因中的促凋亡基因Bax作用于线粒体膜表面,使其通透性改变,Cyt c和凋亡蛋白酶激活因子(Apaf)释放,凋亡小体形成,激活caspases家族,导致细胞凋亡的发生。细胞凋亡与否主要是细胞内的促凋亡基因Bax和抑制凋亡基因Bcl-2相互作用,Bcl-2占优势时细胞存活,Bax占优势时细胞走向凋亡[9]。本实验结果显示:肺IR时, Bcl-2基因及蛋白表达下降,Bax基因及蛋白表达均升高,且Bcl-2/Bax比值下降;AI与Bax及Bcl-2/Bax基因及蛋白表达呈正相关,与Bcl-2基因及蛋白表达呈负相关,这与文献[9]报道一致;而肺IPO中,Bcl-2基因及蛋白表达上调,Bax基因及蛋白表达下降,AI减少,超微结构下肺泡I I型上皮细胞胞膜及细胞器结构完整,线粒体未发生水肿,线粒体脊清晰可见,结果表明了肺IPO可减轻肺IR的细胞损伤和细胞凋亡。
更多的研究表明MAPKs家族在肺IR及其所致的细胞凋亡中发挥了不可忽视的作用[10-11],而ERK1/2是MAPK家族的重要成员,目前ERK1/2的激活在细胞凋亡中究竟发挥何种调节机制仍有争论,更多的研究认为适度激活ERK1/2可以抑制凋亡的发生[12-14]。本室先前研究表明,肺IR时,抑制ERK1/2的激活,导致细胞凋亡率明显增加[8]。在心脏等器官IR中,ERKs被认为有抗凋亡作用,对IR脏器有保护作用[15]。Darling等[4]研究表明,在多种在体和离体的心肌IR中,IPO可通过ERK1/2的激活来减轻IR引起的心肌结构损伤和细胞凋亡。本实验也发现,当应用ERK1/2特异性抑制剂U0126后,IPO中凋亡细胞数增多,即AI值增加,凋亡基因Bax占优势,说明阻断ERK1/2后,IPO对IR的保护作用减弱。应用U0126后,Bax与Bcl-2也发生了相应的改变,这说明ERK1/ 2在内源性凋亡途径中处于Bcl-2/Bax的上游,很可能ERK1/2的激活促使Bcl-2/Bax发生相应的改变,从而减少细胞凋亡。
综上所述,IPO可激活肺组织ERK1/2,使抗凋亡基因Bcl-2表达增加,而促凋亡基因Bax表达减弱,减轻了IR引起的肺组织结构损伤和细胞凋亡。
[1]Ng CS, Wan S, Yim AP. Pulmonary ischaemia-reperfusion injury: role of apoptosis[J]. Eur Respir J, 2005, 25(2): 356-363.
[2]李奎, 白育庭. 单次与多次缺血后处理对肺缺血再灌注损伤的实验研究[J]. 临床外科杂志, 2008, 16(5): 341-343.
[3]Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome[J]. Science, 2002, 298(5600): 1912-1934.
[4]Darling CE, Jiang R, Maynard M, et al. Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK1/2[J]. Am J Physiol Heart Circ Physiol, 2005, 289(4): 1618-1626.
[5]Irving EA, Bamford M. Role of mitogen-and stress-activated kinases in ischemic injury[J]. J Cereb Blood Flow Metab, 2002, 22(6): 631-647.
[6]赵珊, 刘亚坤, 王万铁, 等. 缺血后不同再灌注时间点C57BL/ 6J小鼠肺未折叠蛋白反应的变化及其意义[J]. 温州医学院学报, 2013, 43(2): 84-89.
[7]邱晓晓, 宋张娟, 王万铁, 等. 三七总皂苷对肺缺血/再灌注损伤时细胞凋亡及c-Jun氨基末端激酶的影响[J]. 生理学报, 2012, 64(2): 135-141.
[8]洪加林, 郑艳蓉, 王万铁, 等. 细胞外信号调节激酶通路对大鼠再灌注损伤肺细胞凋亡的影响[J]. 中国临床药理学与治疗学杂志, 2009, 14(10): 1137-1141.
[9]王彤, 刘存志, 刘玉珍, 等. Bcl-2/Bax基因调控机体细胞凋亡的机制研究进展[J]. 中国老年学杂志, 2008, 28(16): 1658-1660.
[10]Zhang X, Bedard EL, Potter R, et al. Mitogen-activated protein kinases regulate HO-1 gene transcription after ischemia-reperfusion lung injury[J]. Am J Physiol Lung CellMol Physiol, 2002, 283(4): L815-L829.
[11]王文, 任玲, 王建楠. MAPK信号通路与细胞凋亡的关系[J].中国实用医药, 2010, 5(15): 260-261.
[12]Hill M, Wernig A, Goldspink G. Muscle satellite (stem) cell, activation during local tissue injury and repair[J]. J Anat, 2003, 203(1): 89-99.
[13]Lee do Y, Lee MW, Lee HJ, et al. ERK1/2 activation attenuates TRAIL-induced apoptosis through the regulation of mitochondria-dependent pathway[J]. Toxicol in Vitro, 2006, 20(6): 816-823.
[14]Gonzalez-Zulueta M, Feldman AB, Klesse LJ, et al. Requirement for nitric oxide activation of p21 (ras)/extracellular regulated kinase in neuronal ischemic preconditioning[J]. Proc Natl Acad Sci USA, 2000, 97(1): 436-441.
[15]Abe Ji, Baines CP, Berk BC. Role of mitogen-activated protein kinases in ischemia and reperfusion injury: the good and the bad[J]. Circ Res, 2000, 86(6): 607-609.
(本文编辑:吴健敏)
Effects of ERK1/2 on pneumocyte apoptosis after lung ischemia/reperfusion injury and ischemic postconditioning
intervention
CHEN Haie, MA Yingchun, HUANG Linjing, HE Jinbo, SONG Dong, ZHENG Mengxiao, YING
Lei, WANG Wantie.Department of Pathophysiology, Ischemia/Reperfusion Injury Research Institute, Wenzhou Medical University, Wenzhou, 325035
Objective: To investigate the role of ERK1/2 on pneumocyte apoptosis after lung ischemia/ reperfusion injury and ischemic postconditioning (IPO) intervention.Methods:Forty adult male Sprague-Dawley rats were randomly divided into 5 groups based upon the intervention (n=8): control group (C), IR group (IR), IR+IPO group (IPO), IPO+solution countrol group (D), IPO+U0126 group (U). Left lung tissue was isolated after the 2 hours of reperfusion, The ultrastructure of the left lung were observed under electron transmission microscopes. Apoptosis index (AI) of lung tissue was determined by terminal deoxynuleotidyl transferase mediated dUTP nick end and labeling (TUNEL) method. The mRNA expression and protein levels of Bcl-2 and Bax were measured by RT-PCR and quantitative immunohistochemistry (IHC).Results:Compared with C group, AI and the expression of Bax of IR were significantly increased, the expression of Bcl-2 and Bcl-2/Bax were significantly decreased (P< 0.05), and ultrastructure abnormality was obviously found in lung tissue. Compared with IR group, all the indexes of IPO except for the expression of Bcl-2 and Bcl-2/Bax were obviously reduced, the expression of Bcl-2 and Bcl-2/Bax was increased (P<0.05). All the indexes between D and IPO were little or no significant (P>0.05). the expression of Bcl-2 and Bcl-2/Bax of U was significantly decreased and other indexes were increased than those of IPO (P<0.05).Conclusion:IPO may attenuate pneumocyte apoptosis in LIRI by activation of ERK1/2 MAPK, up-regulating expression of Bcl-2/Bax ratio.
ischemia/reperfusion; ischemic postconditioning; lung; cell apoptosis; ERK1/2; U0126; Bcl-2; Bax; rats
R363
A
1000-2138(2014)02-0095-06
2013-09-22
温州市高层次人才创新技术重点资助项目(2 01 1-05);浙江省中医药重点学科建设计划项目(2 01 2-X K-A 28)。
陈海娥(1986-),女,河北秦皇岛人,硕士生。
王万铁,教授,博士生导师,Email:wwt@wzmc.edu.cn。
