肾小球疾病患者尿液比较蛋白质组学研究方法的建立
2014-03-21郑春霞高友鹤刘志红
王 玉 孙 伟 左 科 王 霞 郑春霞 高友鹤 刘志红
尿液一直是寻找肾脏病无创性标志物的良好来源。但是,在疾病状态下,肾小球滤过屏障及相关肾单位结构受损,大量血浆中的高丰度蛋白进入尿液,其所形成的高丰度抑制使得尿液蛋白质组学对这类患者的探索面临不少困难。
多种蛋白质组学技术已经应用于这类疾病尿液蛋白的研究,例如二维电泳-质谱(2DE-MS),表面增强激光解吸离子化飞行时间质谱(SELDI-MS),和毛细管电泳-质谱(CE-MS)[1-3]。同位素相对标记与绝对定量技术(iTRAQ)标记的液质联用的蛋白质组学研究方法是一种新的定量蛋白质组学研究策略,它具有定量较准确,全面分析中低丰度蛋白等优点。目前已广泛应用于血液、组织、正常尿蛋白质组学[4-6],但还未应用于肾小球疾病患者尿液的报道。
膜性肾病(MN)和局灶节段性肾小球硬化(FSGS)是临床上导致肾病综合征最常见的两种病理类型。目前对上述疾病的诊断依赖于肾活检组织病理学检查,尚缺乏用于诊断、鉴别诊断、判断病情及指导治疗的无创性标志物。本研究中,我们建立了联合白蛋白/IgG抗体清除和ITRAQ标记的二维液相色谱与串联质谱联用技术(2D-LC MS/MS)用的尿液蛋白质组学检测方法,并对临床表现肾病综合征的MN和FSGS患者的尿液蛋白质组学进行比较分析。
对象和方法
研究材料本研究得到南京军区南京总医院伦理委员会批准。尿液样本来源于2012年南京军区南京总医院肾脏科住院和门诊患者,标本采集经患者本人或家属知情同意。收集研究对象的随机尿50 ml,立即放入4℃暂存。
研究对象入选标准:临床表现肾病综合征,病理证实MN或FSGS,目前肾病范围蛋白尿未缓解(尿蛋白定量>3.5 g/d,血清白蛋白<30 g/L)。排除标准:(1)所有患者排除代谢性疾病、自身免疫病、肿瘤、重金属中毒等继发性因素;(2)排除收集尿液时合并感染、血栓、血清肌酐升高(>176.8 μmol/L)、严重肝功能不全的患者;(3)排除月经期女性。
研究对象随机分组:MN分为三组, 每组5例患者(MN1, MN2 和MN3组); FSGS分为三组,每组5例患者(FSGS1, FSGS2 和FSFS3 组)(表1)。
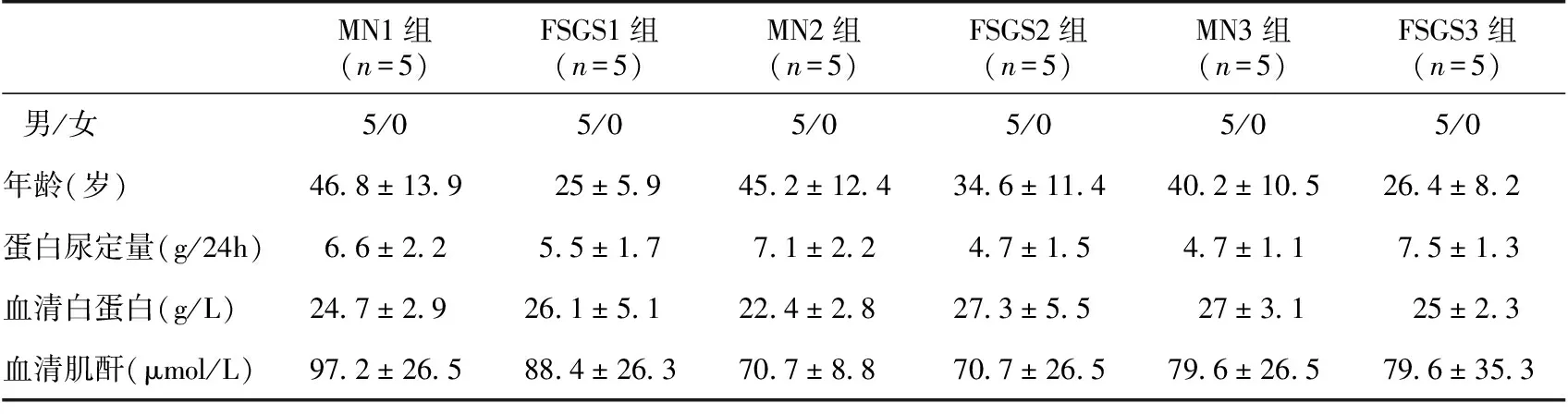
表1 膜性肾病(MN)和局灶节段性肾小球硬化(FSGS)患者的临床资料及分组
试剂和仪器本实验所用二硫苏糖醇(DTT)、碘乙酰胺(IAA)、吗啡啉乙磺酸(MES)、碳酸氢铵等购自Sigma公司,乙腈、甲酸、三氟乙酸等购自ABI公司,ProteoPrep免疫亲和白蛋白/IgG清除试剂盒购自Sigma公司,10 kd滤器购自Millipore公司,胰酶购自Protégé公司,C18 spin column除盐柱购自The Nest Group公司,iTRAQ试剂购自ABI公司,真空离心浓缩仪SPD121P-230购自Thermo公司,Magic C18AQ液相色谱柱购自Michrom公司,LTQ-Obitrap质谱仪购自thermo公司。
样品处理
丙酮沉淀尿液样本于5 000g,4℃,离心30 min 去除细胞碎屑,加入3倍体积-20℃预冷丙酮,4℃放置4h时,14 000g,4℃,离心30 min,去除上清,保留蛋白沉淀。碳酸氢铵复溶蛋白沉淀,10 000g,4℃,离心10 min去除沉淀,保留上清,Bradford法测定蛋白浓度。
蛋白质样品组内混合从MN1、MN2、MN3、FSGS1、FSGS2、FSGS3组内的各5例样品取等量蛋白进行组内混合。
去除高丰度蛋白按照ProteoPrep免疫亲和白蛋白/IgG清除试剂盒操作说明,去除以上各组高丰度蛋白,Bradford法测定蛋白浓度。
蛋白质酶切各组样品中分别加入20 mmol/L DTT 37℃还原1h,再加入50 mmol/L IAA室温避光反应45 min,加入10 kD膜离心,8 mol/L尿素-Tris/HCl溶液清洗2次,碳酸氢铵清洗2次后复溶,按1∶20加入胰酶37℃消化过夜,多肽样品除盐后抽干备用。
iTRAQ标记分别向各组抽干后肽段中加入等量iTRAQ试剂盒自带试剂溶解肽段,分别取等量6组溶解后肽段进行混合,作为内参;取两组iTRAQ试剂,向iTRAQ标记试剂114、115、116、117中分别加入等量70%乙醇重悬标记试剂;向一组iTRAQ试剂114、115、116、117重悬试剂中加入内参、MN1、MN2、MN3;另一组iTRAQ试剂114、115、116、117重悬试剂中加入内参、FSGS1、FSGS2、FSGS3,室温孵育1h;取标记后的内参(114标记)、MN1(115标记)、MN2(116标记)、MN3(117标记);内参(114标记)、FSGS1(115标记)、FSGS2(116标记)、FSGS3(117标记)样品按1∶1∶1∶1分两组进行混合,真空离心浓缩抽干样品,保存于-20℃。实验前将样品溶于适量1‰氨水(pH=10)中,浓度调整到5 μg/ul。
质谱分析
高pH值反相液相色谱分离反相柱为250 mm×4.6 mm,洗脱液为5%~30%缓冲液A(0.1%氨水,99.9%乙腈,pH=10,流速1 ml/min),洗脱时间为60 min。每1 min收集1个组分,共收集60个组分。各组分抽干后,每间隔2个组分混合,共混合成20个组分,进行液质联用分析。
液质联用分析反相分离:反相柱为100 mm×0.075 mm,洗脱液为5%~30%缓冲液B(0.1%甲酸,99.9%乙腈,流速300 nl/min),洗脱时间为2h。TripleTOF5600质谱检测:质核比检测范围为350~1 250 amu,应用数据依赖方式进行二级质谱扫描,每次全扫描后做30次二级扫描,母离子质核比宽度为0.7 amu,35%标准碰撞能量,动态排除时间为15s。
生物信息学分析质谱数据用MASCOT(V2.3.02)进行检索,所用数据库为swissProt人的蛋白质数据库。参数:酶为Trypsin,误切位点2,多肽和子离子质量误差范围为0.05 D,半胱氨酸的脲甲基化修饰。所有质谱检测结果同时应用反相数据库检索方法评估数据的假阳性率(假阳性率=反相数据库鉴定肽段数/正向数据库鉴定肽段数×100%)。检索结果用Scafford软件进行分析。蛋白和多肽假阳性率<1%,每个蛋白至少有2个多肽。
Gene Ontology(GO)分析应用PANTHER软件 (http://www.pantherdb.org/)根据分子功能、生物过程、细胞定位三种注释进行蛋白质分类。
结 果
蛋白尿蛋白质组学图谱鉴定通过白蛋白/IgG抗体清除结合iTRAQ标记2D-LC MS/MS分析(图1),在6组蛋白尿样品中共鉴定到809个蛋白。根据谱图数相对定量,估算鉴定蛋白中丰度排名前10位的蛋白,并与血蛋白质组前10位高丰度蛋白进行比较[7]。蛋白尿蛋白质组前10位蛋白都来自于血液,但蛋白尿前10位蛋白与血蛋白质前10位蛋白种类及排序差别较大。
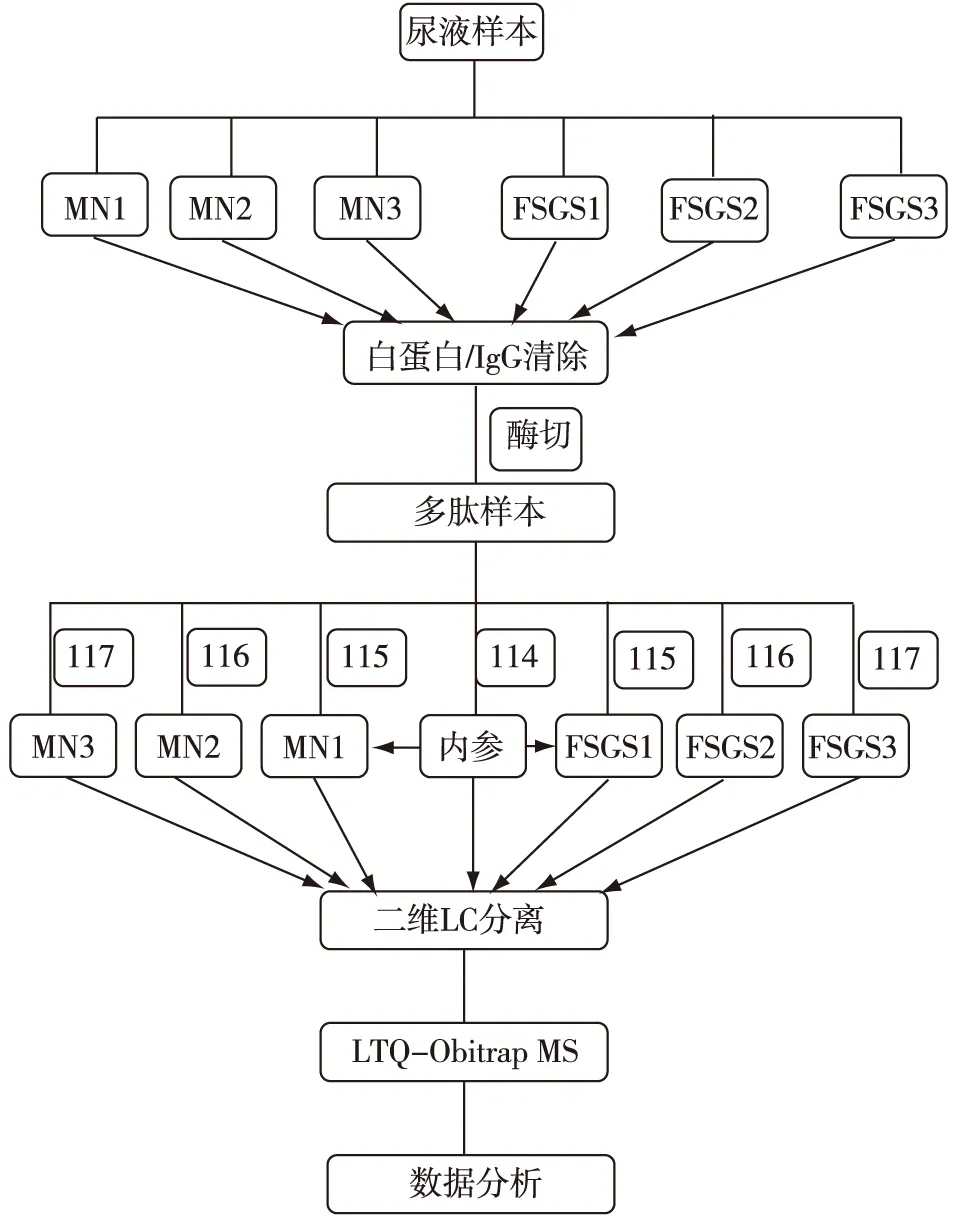
图1 实验流程图
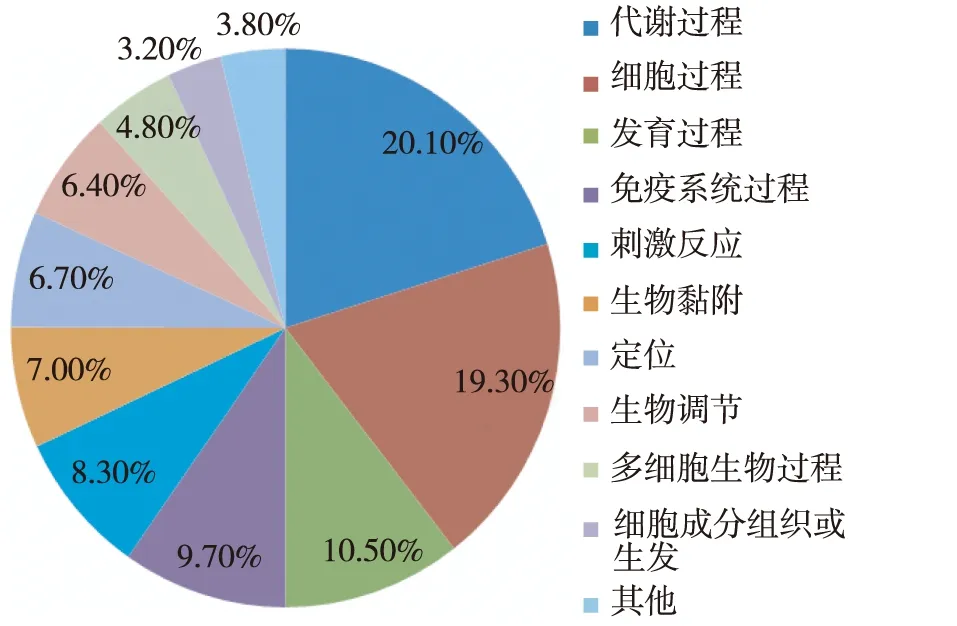
图2 根据蛋白质参与的生物学过程进行分析的结果
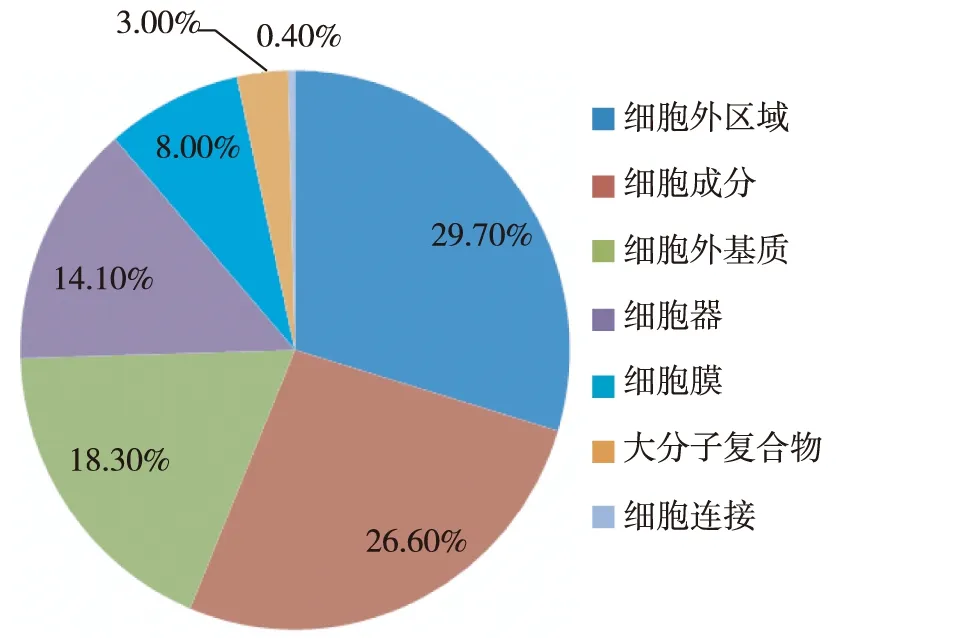
图3 根据蛋白质所属的细胞成分进行分析的结果
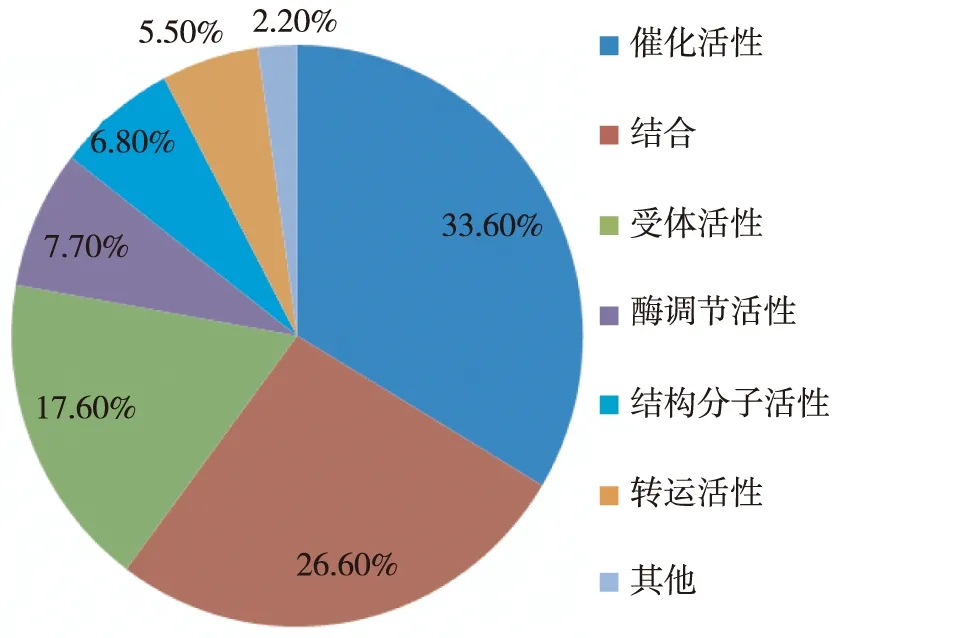
图4 根据蛋白质的分子功能进行分析的结果
GO功能分析应用PANTHER软件对所有鉴定蛋白质进行注释分类,显示根据分子功能,前三位分类蛋白质属于催化活性、结合、受体活性(图2)。根据生物过程,前三位分类蛋白属于代谢过程、细胞过程、发育过程(图3)。根据细胞定位,前三位分类蛋白质属于细胞外区域、细胞成分、细胞外基质(图4)。
差异蛋白筛选本实验采用两组4标 iTRAQ试剂,每组iTRAQ试剂有114、115、116、117四种同位素。一组iTRAQ试剂114、115、116、117分别标注内参、MN1、MN2、MN3,另一组iTRAQ试剂114、115、116、117分别标注内参、FSGS1、FSGS2、FSGS3。共进行了3次重复LC-MS/MS质谱鉴定,以报告离子为m/z 114作内参进行相对定量,分别计算每个蛋白在MN和FSGS三次质谱鉴定中的115∶114,116∶114,117∶114比值,即可获得MN1、MN2、MN3、FSGS1、FSGS2、FSGS3共6个相对定量值,根据115标注MN1/FSGS1,116标注MN2/FSGS2,117标注MN3/FSGS3比值即可得到三组每个蛋白在MN和FSGS丰度的相对变化值fold chang,根据115、116、117三个值可计算每个蛋白在MN或FSGS三次重复实验的变异系数CV1、CV2,根据3个fold change计算变异系数CV3。符合以下标准进入差异蛋白分析:(1)鉴定蛋白特异性多肽≥2;(2) 任一fold chang≥1.5;(3)CV1、CV2、CV3值均<0.6。符合条件差异蛋白16个,具体与疾病的关系仍待进一步的研究加以确认。
讨 论
蛋白尿不仅是肾脏病最具诊断意义的变化,其程度也是临床上用于判断病情、评估治疗效果和预测预后的一个重要指标。目前,临床上只能对其进行定量分析,缺乏质的分析,更缺乏疾病特异性的相关蛋白质标志物。利用蛋白质组学的方法,从中患者尿液中寻找肾脏疾病标志物将有助于我们提高对这类患者诊断和治疗的水平。
我们在研究中发现,大量蛋白尿蛋白质组有着接近于血浆蛋白质组的高丰度抑制。怎样减少蛋白质组研究中的高丰度抑制效应,使蛋白质组中、低丰度蛋白成分的检测不至于因此而受到干扰是目前蛋白质组学研究中一个极具挑战的问题。尽管,采用蛋白质分离联合或不联合高丰度蛋白清除技术已广泛用于蛋白质组学的研究,但是,将其用于肾脏病患者尿液蛋白质组学的研究还很少。
六肽库是基于20 种天然氨基酸合成的六肽配体组合文库,理论上可与近乎无限种类的氨基酸残基结合,能结合每种多肽的配体是有限的,一旦六肽饱和,过量的蛋白就会被冲洗掉,六肽库在有效降低高丰度蛋白的同时,相对富集了低丰度蛋白。该技术在人类正常尿液、血液、组织样本中得到了应用[8-10],提高了蛋白的鉴定数目。抗体亲和法是利用几种单克隆或多克隆抗体特异性清除样本中的高丰度蛋白,已有同时清除2种、6种、14种血液高丰度蛋白的抗体组合试剂盒,这些试剂盒被广泛应用于寻找血、尿蛋白质组标志物[11-13]。日本学者提出差异溶解术,利用有机溶剂溶取小分子量蛋白的特性,能较好的获取分析血浆中的低丰度蛋白,但该技术并不是基于全蛋白质组,目前该技术应用尚少[14]。Chen等[12]将能清除14种高丰度蛋白的抗体柱与六肽库分别应用于正常尿蛋白质组,通过iTRAQ技术比较二者的重复性发现,抗体柱的重复性较六肽库好。 我们的前期实验比较了白蛋白/IgG抗体清除、六肽库、差异溶解术三种高丰度清除策略应用于肾病水平蛋白尿蛋白质组的效果,发现经六肽库处理后样品蛋白鉴定数略多于白蛋白/IgG抗体清除、差异溶解,但白蛋白/IgG抗体清除鉴定重复性明显好于六肽库和差异溶解(数据未发表),可能更适合比较蛋白质组学研究。目前蛋白质组学常用的蛋白质分离技术有二维电泳(2DE)和高效液相色谱(HPLC)。2DE由于其耗时费力、解析度及重复性较差等缺陷,目前应用已越来越少。而HPLC因其适用性广等优点,目前在蛋白质组学研究中,采用液相色谱与质谱联用的越来越多。目前基于质谱的蛋白质定量方法有非标记定量和稳定同位素标记定量,非标记定量法所需样品量少、操作简单,但是一次只能分析一个样品,且在高丰度蛋白存在时,定量可能不准确;相比之下,同位素标记方法可分析多个样品,定量更准确[15]。目前应用较普遍的同位素标记方法是iTRAQ技术[16]。
本研究应用白蛋白/IgG抗体清除联合iTRAQ标记的2D-LC MS/MS对临床表现肾病综合征的MN和FSGS患者的尿液进行比较蛋白质组学,从患者的蛋白尿中,我们共鉴定出809个蛋白,这是目前针对蛋白尿的最大规模的蛋白质图谱研究。GO分析显示根据分子功能,鉴定蛋白主要属于催化活性、结合、受体活性三个类别。根据生物学活性,鉴定蛋白主要参与代谢、细胞活动和发展调控三个类别。根据细胞定位,鉴定蛋白主要属于细胞外区域、细胞成分、细胞外基质三个类别。通过MN和FSGS患者尿液蛋白质谱图数进行相对定量,我们发现蛋白尿中前10位蛋白与血浆前10位蛋白差别很大,提示即使在病理情况下,肾脏可能对不同种类的蛋白仍保持不同的处理功能,蛋白尿可能有着不同于血蛋白质组的组成架构。目前仍无针对蛋白尿高丰度蛋白进行抗体清除的检测试剂盒,对蛋白尿整体构架了解较少可能是制约其发展的重要原因。我们对蛋白尿的较全面的图谱分析及根据谱图数相对定量获得的蛋白相对丰度构成可能为今后肾脏病尿液蛋白质组学研究提供了基础。通过iTRAQ标记定量,我们筛选了16个差异蛋白,他们的生物学功能分别包括参与纤溶、免疫反应、代谢过程、细胞增生等。
小结:本研究联用高丰度蛋白抗体清除及iTRAQ标记的2D-LC MS/MS对大量蛋白尿蛋白质组进行分析,得到较全面尿液蛋白质组学图谱及一系列在MN和FSGS差异表达的蛋白质,表明该技术在大量蛋白尿尿液蛋白质组学研究上可能有较好的应用前景。研究中获得的尿液蛋白质组学图谱为开展伴大量蛋白尿肾脏病患者尿液蛋白质组学研究提供了基础;从MN和FSGS患者尿液中获得的差异蛋白有待进一步验证其与疾病诊断、治疗及预后的关系。
1Somparn P,Hirankarn N,Leelahavanichkul A,et al.Urinary proteomics revealed prostaglandin H2D-isomerase,not Zn-α2-glycoprotein,as a biomarker for active lupus nephritis.J Proteomics,2012,75(11):3240-3247.
2Zhang X,Jin M,Wu H,et al.Biomarkers of lupus nephritis determined by serial urine proteomics.Kidney Int,2008,74(6):799-807.
3Rossing K,Mischak H,Dakna M,et al.Urinary Proteomics in Diabetes and CKD.J Am Soc Nephrol,2008,19(7):1283-1290.
4Sigdel TK,Salomonis N,Nicora CD,et al.The identification of novel potential injury mechanisms and candidate biomarkers in renal allograft rejection by quantitative proteomics.Mol Cell Proteomics,2014,13(2):621-631.
5Yang LS,Xu XE,Liu XP,et al.iTRAQ-based quantitative proteomic analysis for identification of oligodendroglioma biomarkers related with loss of heterozygosity on chromosomal arm 1p.J Proteomics,2012,77:480-491.
6Gautam P,Nair SC,Gupta MK,et al.Proteins with altered levels in plasma from glioblastoma patients as revealed by iTRAQ-based quantitative proteomic analysis.PLoS One,2012,7(9):e46153.
7Farrah T,Deutsch EW,Omenn GS,et al.A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas.Mol Cell Proteomics,2011,10(9):M110.006353.
8Castagna A,Cecconi D,Sennels L, et al.Exploring the hidden human urinary proteome via ligand library beads.J Proteome Res,2005,4(6):1917-1930.
9Guerrier L,Claverol S,Fortis F,et al.Exploring the platelet proteome via combinatorial,hexapeptide ligand libraries.J Proteome Res,2007,6(11):4290-4303.
10 Hartwig S,Czibere A,Kotzka J,et al.Combinatorial hexapeptide ligand libraries (ProteoMiner):an innovative fractionation tool for differential quantitative clinical proteomics.Arch Physiol Biochem,2009,115(3):155-160.
11 Mebazaa A,Vanpoucke G,Thomas G,et al.Unbiased plasma proteomics for novel diagnostic biomarkers in cardiovascular disease:identification of quiescin Q6 as a candidate biomarker of acutely decompensated heart failure.Eur Heart J,2012,33(18):2317-2324.
12 Chen CL,Lin TS,Tsai CH,et al.Identification of potential bladder cancer markers in urine by abundant-protein depletion coupled with quantitative proteomics.J Proteomics,2013,85:28-43.
13 Ma Y,Peng J,Huang L,et al.Searching for serum tumor markers for colorectal cancer using a 2-D DIGE approach.Electrophoresis,2009,30(15):2591-2599.
14 Kawashima Y,Fukutomi T,Tomonaga T,et al.High-yield peptide-extraction method for the discovery of subnanomolar biomarkers from small serum samples.J Proteome Res,2010,9(4):1694-1705.
15 Domon B1,Aebersold R.Options and considerations when selecting a quantitative proteomics strategy.Nat Biotechnol,2010,28(7):710-721.
16 Ross PL1,Huang YN,Marchese JN,et al.Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents.Mol Cell Proteomics,2004,3(12):1154-1169.
