优化易错PCR条件以提高毕赤酵母GAP启动子文库突变效率
2014-03-17秦秀林钱江潮储炬
秦秀林钱江潮储炬
(1.广西大学生命科学与技术学院,南宁 530004;2.华东理工大学生物反应器工程国家重点实验室,上海 200237)
优化易错PCR条件以提高毕赤酵母GAP启动子文库突变效率
秦秀林1钱江潮2储炬2
(1.广西大学生命科学与技术学院,南宁 530004;2.华东理工大学生物反应器工程国家重点实验室,上海 200237)
高效的随机突变策略对于构建含有丰富突变体的启动子文库至关重要。为了建立一个突变率适中并能获得较多有益突变子的易错PCR(error-prone PCR,EP-PCR)条件,实现毕赤酵母GAP启动子的高效突变,对EP-PCR反应条件进行了优化。考察了模板浓度、反应循环数和Mg2+浓度对EP-PCR的产物得率和突变率的影响后,确定了适于GAP启动子突变的EP-PCR条件:模板浓度、反应循环数和Mg2+浓度分别为1 ng/μL、25和10 mmol/L。优化EP-PCR条件后,GAP启动子突变率为1.1%,连续进行3轮EP-PCR后突变率可达到4.0%。利用优化后EP-PCR对GAP启动子进行随机突变,筛选了250个突变子,获得5个启动子强度高于野生型GAP启动子的突变体,有益突变达到了2%,可用于构建GAP启动子文库。
易错PCR 突变效率 随机突变 毕赤酵母 GAP启动子
利用易错PCR(Error prone PCR,EP-PCR)或DNA改组(DNA shuffling)等基因突变技术对基因启动子区进行改造构建启动子文库,并已在原核和真核微生物的代谢工程、功能基因组学和合成生物学中得到了成功应用[1-3]。启动子文库是不同强度的启动子集合体,在构建时,对天然启动子序列进行突变,再从中筛选出合适强度的多个突变启动子从而组成启动子文库[4]。由于启动子的碱基序列直接影响其调控基因转录的强度,故只要所构建的文库包含足够丰富的突变序列,筛选量足够大,就可以从中筛选出一系列具有不同调控强度的突变启动子,构成能对其下游基因实施精确微调的文库。
近年来,EP-PCR在启动子文库构建过程中应用非常广泛[5-7]。Alper等[4]通过EP-PCR获得了噬菌体PL-λ启动子活性变化范围较大的突变启动子文库,启动子的活性在196倍的范围内变化,而报告基因gfp的转录分析证明突变启动子调控的转录水平可在 325倍的范围内变化,证明可通过随机突变获得启动子活性差别较大的突变体,以达到对基因进行精细调节的目的。Nevoigt等[8]通过筛选EP-PCR突变的溶氧敏感型启动子DAN1突变体文库,获得了在微氧条件下也能够诱导表达的启动子突变体,其启动子活性在微氧条件下是野生型启动子的1.8-2.9倍。Hartner等(Hartner et al. 2008)对巴斯德毕赤酵母强启动子AOX1的转录因子结合序列(TFBSs)进行突变改造后,利用96孔深孔板进行高通量筛选,挑选出的突变启动子的活性可在原AOX1启动子强度6%至160%之间变化。启动子文库能实现目的基因表达的精确调控,但其不足之处是突变后获得的有益突变体较少,Nevoigt等[8]在筛选了12 000个转化子后只有6个突变体的启动子活性较野生型启动子提高;Alper等[4]在筛选过程中从30 000个菌落中只筛到不足0.1%具有有用启动子的菌株。因此,启动子文库构建的主要难点不是繁重的高通量筛选,而是建立一个能获得较多有益突变的EP-PCR条件。
作为真核生物,毕赤酵母具有高等真核表达系统的许多优点:如蛋白加工、折叠、翻译后修饰等,并已成功用于异源蛋白和代谢物的表达。这些使得毕赤酵母广泛地应用于基础研究和大规模工业化生产。利用EP-PCR对毕赤酵母组成型三磷酸甘油醛脱氢酶启动子GAP进行随机突变,构建GAP突变文库可以为毕赤酵母基因的连续表达调控提供一新的基因操作平台。EP-PCR的关键在于选择适当的突变频率,一般有益突变频率较低,绝大多数突变是有害或中性。若突变频率过高,几乎无法筛选到有益突变;若突变频率太低,则未发生任何突变的野生型在突变群体中将占优势,也很难筛选到理想突变体;而目标基因的突变率在1%-5%范围时才能获得较多的有益突变达到理想的诱变结果[9,10]。
因此,为了获得大量的具有丰富突变的GAP启动子并增加启动子的突变效率,本研究对EP-PCR反应条件:模板浓度、循环数和Mg2+浓度分别进行优化,并应用优化后条件对GAP启动子进行突变。
1 材料与方法
1.1 材料
1.1.1 试验材料 大肠杆菌DH5α由本实验室保藏,毕赤酵母菌株GS115和pGAP Z载体购自Invitrogen公司,克隆载体pMD18-T购自TaKaRa公司。DNA序列经TaKaRa公司测定。
1.1.2 试剂 脱氧核糖核苷酸(dATP、dTTP、dGTP和dCTP)、TaqDNA 聚合酶均购自上海生工生物工程技术服务有限公司。限制性内切酶和连接反应试剂盒均为TaKaRa公司产品,质粒小量抽提、胶回收试剂盒和PCR反应液纯化试剂盒为AxyGen公司产品。其他化学试剂均为国产分析纯。
1.2 方法
1.2.1 菌株的培养 大肠杆菌DH5α在LLB培养基中37℃培养,转化重组质粒后LLB培养基需添加zeocin 至终浓度为25 μg/mL。毕赤酵母菌株在YPD培养基(1% yeast extract,2% peptone,2% glucose)、BMD培养基(1.34% YNB,1% glucose,4×10-5% biotin,100 mmol/L potassium phosphate,pH6)或 BMDY培养基(1% yeast extract,2% peptone,1.34% YNB,1%葡萄糖,4×10-5% biotin,100 mmol/L potassium phosphate,pH 6)中28℃培养。
1.2.2 毕赤酵母GS115的转化 重组质粒经BspEI线性化后电转化毕赤酵母GS115感受态细胞,用引物GAP primer和GFP-r(表1)经菌落PCR确定重组基因整合的正确性。转化子的筛选用含0.4 μg/mL biotin 的MD平板(1.34%YNB,2%葡萄糖,1.5%琼脂粉)。

表1 引物列表
1.2.3 EP-PCR突变GAP启动子 以质粒pGAP Z
为模板,用引物GAP-EPf /GAP-EPr(表1)经EPPCR获得突变的GAP启动子,EP-PCR反应体系为100 μL,所含Mn2+、dATP、dGTP、dCTP、dTTP浓度固定不变分别为0.5、0.2、0.2、1和1 mmol/L。10×PCR缓冲液:100 mmol/L Tri-HCl(pH8.3),500 mmol/L KCl,20 mmol/L MgCl2。易错PCR反应条件:94℃预变性3 min;94℃变性1 min,54℃退火1 min,72℃延伸1 min,25个循环。
1.2.4 GAP启动子突变文库的筛选 GAP启动子突变文库高通量筛选采用48深孔板,每孔装900 μL培养基,于30℃,220 r/min培养。筛选方式参照之前报道的方法[11]:将转化后的转化子涂布于MD筛选平板,待单菌落直径至3 mmol/L左右接种至保种板培养(Master plate)培养;取保种板24 h培养菌液至对应的预培养板孔(preculture plate)中培养,预培养板的48深孔板每孔装900 μL的BMD(0.2%葡萄糖);从预培养板取培养48 h的菌液至主培养板(Main culture plate)中对应孔中,主培养板的48深孔板每孔装900 μL的BMD(1%葡萄糖),培养36 h后取菌液测定菌体浓度和yEGFP表达强度。
1.2.5 菌体密度测定 菌液稀释一定倍数后在波长600 nm处以无菌培养基为对照进行比色,OD600=OD读数×稀释倍数。
1.2.6 重组菌中yEGFP荧光强度检测 将主培养板培养了36 h的菌液取30 μL至装有220 μL PBS的96孔板中,利用多功能酶标仪((BioTek Synergy 2)检测酵母增强型绿色荧光蛋白yEGFP荧光强度(激发波长:395 nm,发射波长:509 nm)。检测yEGFP荧光强度时,以不表达yEGFP的基因工程菌G/GH[11]作为对照去除背景干扰。比荧光强度F/OD600(RFU/ OD600)为荧光强度值比上对应细胞密度OD600。
2 结果
2.1 EP-PCR反应体系中模板浓度的优化
为了提高目的基因的突变率并保证能获得较多的扩增产物,首先对EP-PCR反应体系中模板(pGAP Z质粒)浓度进行了优化。反应体系中模板终浓度为0.1、0.5、1和2 ng/μL时EP-PCR获得的产物(图1)经纯化后利用NanoDrop分别测定其DNA的浓度。模板浓度为0.1 ng/μL和0.5 ng/μL时,EP-PCR产物浓度分别只达到25 ng/μL和40 ng/μL;当模板浓度达到1 ng/μL时,EP-PCR产物浓度增加到85 ng/μL;增加模板浓度至2 ng/μL时,EP-PCR产物量没有显著提高(90 ng/μL)。因此,EP-PCR反应体系中模板浓度为1 ng/μL时,可以获得较多的产物。
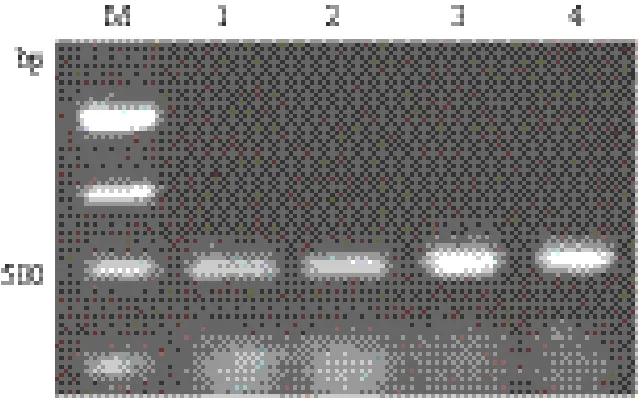
图1 反应体系中模板浓度对EP-PCR的影响
2.2 EP-PCR反应体系循环数的优化
为了研究循环数对EP-PCR产物浓度的影响,将EP-PCR反应体系中的循环数设为20、25、30和35(反应体系中模板浓度为1 ng/μL),并利用NanoDrop检测对应的产物浓度,结果如图2。当扩增只进行20个循环时,EP-PCR产物浓度约35 ng/μL;循环数增加至25时,产物浓度提高至约85 ng/μL;循环数增加至30和35时,产物浓度分别为83 ng/μL和88 ng/μL。可见达到25个循环后,产物浓度达饱和,增加循环数,并不能提高产物的浓度。因此,EP-PCR反应体系最佳的循环数为25。

图2 循环数对EP-PCR的影响
2.3 EP-PCR反应体系中Mg2+浓度的优化
为了优化易错PCR的反应条件,达到所需要的突变率(1%-5%),将反应体系的模板浓度和循环
数分别设为1 ng/μL和25,以不同的Mg2+浓度:4、6、8 、10、12 和15 mmol/L分别进行了易错PCR反应,结果如图3。Mg2+浓度在4 -10 mmol/L范围内可扩增出单一的目的条带且产物浓度均达到约90 ng/μL;当Mg2+浓度为12 mmol/L时虽然扩增出了目的条带但也出现了较多的非特异性条带;而Mg2+浓度增加至15 mmol/L时产物出现弥散现象,没有扩增出特异性条带。
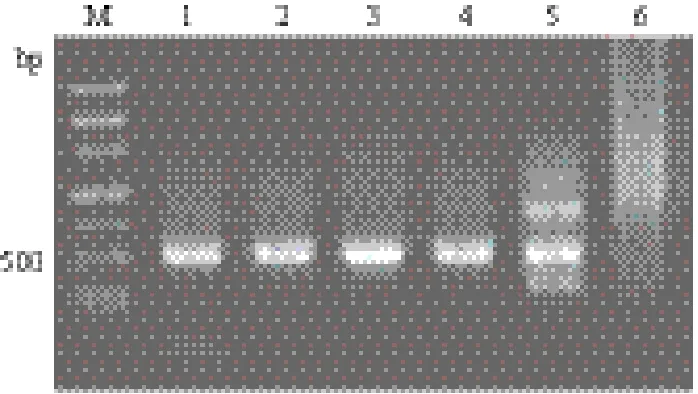
图3 Mg2+浓度对EP-PCR的影响
将各Mg2+浓度反应产物克隆至载体pMD18-T并分别随机挑选10个突变子测序以统计其突变率。结果如表2,Mg2+离子浓度为4 mmol/L时所产生的突变率较低只有0.6%,而Mg2+离子浓度为10 mmol/L时产生的突变率为1.1%。6 mmol/L和8 mmol/L的Mg2+浓度易错PCR反应突变率分别为0.74%和0.83%。这些突变序列与野生型GAP启动子序列相比,突变碱基分布在整个启动子序列,无明显规律。试验表明Mg2+离子浓度为10 mmol/L时产生的突变率较适中能满足试验要求,因此,EP-PCR反应体系的Mg2+离子浓度为10 mmol/L。

表2 不同反应条件易错PCR的突变率
2.4 连续进行多轮EP-PCR以提高突变率
为了增加EP-PCR的突变率,在第1轮EP-PCR的基础上又进行第2或第3轮的EP-PCR。试验中,EP-PCR反应体系的Mg2+、Mn2+、dATP、dGTP、 dCTP、dTTP和模板浓度固定不变分别为10 mmol/L、0.5 mmol/L、0.2 mmol/L、0.2 mmol/L、1 mmol/L、1 mmol/L和1 ng/μL,进行25个循环。第2和第3论EP-PCR的模板分别为第1轮EP-PCR和第2轮EP-PCR的回收产物,模板的添加量为1%(V/V),每轮随机挑选10个突变体进行克隆并测序。经过3轮EP-PCR,突变率可达到1.1%-4.0%(表3)。变启动子的序列各不相同,突变碱基分布在整个启动子序列,无明显规律。连续进行3次EP-PCR突变率为4.0%,达到了预期的突变率,可用于GAP启动子的突变。

表3 经过3轮易错PCR反应后的突变率
2.5 应用优化后EP-PCR对GAP启动子进行突变
为了检测突变启动子强度,构建了突变启动子调控报告基因(酵母增强型绿色荧光蛋白yEGFP基因egfp)表达载体及对应重组菌,通过测定重组菌yEGFP荧光强度以检测对应的突变启动子强度。
首先以质粒pGAP Z为模板,用引物GAPEPf/GAP-EPr(表1)经EP-PCR获得突变的GAP启动子,经限制性内切酶SpeI和NotI双酶切,将突变的GAP启动子混合片段与经SpeI和NotI双酶切后载体pGHg[11]连接,获得重组混合质粒,将这些重组混合质粒用限制性酶BspeI酶切线性化,电转毕赤酵母GS115,获得突变GAP启动子重组毕赤酵母细胞库。转化后涂布MD平板,30℃倒置培养3-5 d,直到单克隆出现。随机挑选30个克隆,采用菌落PCR鉴定转化子,PCR所用引物分别为GAP primer和GFP-r(表1),阳性克隆PCR条带理论大小约800 bp。图4的结果表明,毕赤酵母GAP突变启动子细胞库构建成功。
利用48深孔板,通过高通量筛选方法[11]筛选所构建的突变体,并用荧光酶标仪检测重组菌的yEGFP荧光强度。随机挑取了250个突变子进行筛选,检测到荧光强度较重组菌G/GHg(野生型GAP启动子调控yEGFP表达菌株)显著提高的重组菌
(G/GaHg、G/GbHg、G/GcHg、G/GdHg和G/GeHg)5个,其荧光强度值见表4。为了验证这些重组菌的yEGFP荧光强度改变确实是由于启动子序列突变引起,将突变GAP启动子序列克隆并测序。测序结果表明重组菌中对应的突变启动子Ga、Gb、Gc、Gd和Ge的序列各不相同(图5),与野生型GAP启动子序列相比,突变碱基分布在整个启动子序列,无明显规律。这一结果表明重组菌中的启动子序列确实发生了突变,重组菌yEGFP表达强度不同也确实是由启动子的突变引起,利用优化条件后EP-PCR能高效筛选到有益突变子(启动子强度增强)。

图4 毕赤酵母GAP突变启动子重组菌菌落PCR验证

表4 含突变启动子重组菌的绿色蛋白荧光强度
3 讨论
EP-PCR是一种简便快速地在DNA序列中随机制造突变的方法,而且EP-PCR方法对目的基因没有片段大小的限制,并且因为序列具有几乎100%的均一性,所以得到的克隆在遗传学上比较稳定。与传统的随机突变中常用的亚硝基胍相比,EP-PCR突变获得的突变文库,其性状的多样性远远大于致死率为40%-50%时亚硝基胍的突变效果[12]。然而利用EP-PCR进行随机突变,一般1 kb目标基因内只有1.5-6.6个碱基发生碱基替换(突变率:0.15%-0.66%)[13,14],而目标基因的突变率在1%-5%范围时才能获得较多的有益突变以达到理想的诱变结果[9,10]。因此,为了获得大量的具有丰富突变的启动子并增加启动子的突变效率(1%-5%范围)必须对EP-PCR反应条件进行优化。常规方式是通过改变传统PCR反应体系中某些组分的浓度,如模板、dNTP(不同的脱氧核糖核酸配比浓度)[15]、Mn2+[16]和Mg2+的浓度,使碱基在一定程度上随机错误而创造序列的多样性。其中,Mn2+浓度通常为0.5 mmol/L,而为了避免碱基突变的偏向性dATP∶dGTP∶dCTP∶dTTP的比例通常设为1∶1∶5∶5[17]。因此,本研究中将EP-PCR反应体系的Mn2+、dATP、dGTP、dCTP、dTTP浓度固定不变分别设为0.5、0.2、0.2、1和1 mmol/L,在此条件下,研究了EP-PCR反应中模板浓度、循环数和Mg2+浓度对其影响。
EP-PCR反应体系中模板的初始浓度会影响基因的突变效率和扩增产物的量。反应体系中模板的初始浓度越低,模板所经历的扩增次数越多,最后产物中积累的突变位点越多,即突变效率越高[18];而提高反应体系中模板的初始浓度,突变效率会降低[19]但最终扩增到的产物量会增加。本研究优化EP-PCR反应体系中模板浓度(1 ng/μL)后,在获得较多产物同时保证了目的基因较高的突变率。
在优化EP-PCR反应体系循环数的过程中也发现25个循环得到的产物浓度可达到约85 ng/μL,再增加循环数,产物的浓度并没有增加。这是因为在一定倍增次数范围内,EP-PCR反应过程中目的基因倍增次数与突变效率相关,即目的基因倍增次数越高产生的突变位点越多,同时形成的目的产物浓度也越高;但扩增产物达到一定浓度后(5-50 ng/μL)[20],增加循环数并不能提高产生的量。
很多研究显示,易错PCR的突变率受Taq酶保真性的影响。加入Mn2+后,Taq酶保真性随着反应体系中Mg2+的浓度增加而降低。为了增加EP-PCR的突变率,可以在第1轮EP-PCR的基础上又进行
第2或第3轮的EP-PCR[21]。EP-PCR反应体系的Mg2+离子浓度达到10 mmol/L时GAP启动子产生的突变率达到了1.1%,经过3轮连续EP-PCR,GAP启动子突变率的范围为1.1%-4.0%,符合产生有益突变较多突变率范围(1%-5%)。利用优化后EPPCR对GAP启动子进行突变,通过高通量筛选250个突变子,获得5个启动子强度高于野生型GAP启动子的突变体,有益突变达到了2%。

图5 突变GAP启动子序列比对
4 结论
本研究对EP-PCR反应中模板浓度、循环数和Mg2+浓度分别进行了优化,优化条件后,经过3轮EP-PCR,突变率可达到1.1%-4.0%。利用优化后的EP-PCR反应条件对GAP启动子进行突变,筛选了250个突变子,可挑选出启动子强度较野生型GAP启动子显著提高的突变子5个。
[1] Braatsch S, Helmark S, Kranz H, et al.Escherichia colistrains with promoter libraries constructed by Red/ET recombination pave the way for transcriptional fine-tuning[J].Biotechniques, 2008, 45(3):335-337.
[2] Siegl T, Tokovenko B, Myronovskyi M, et al. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes[J]. Metabolic Engineering, 2013, 19:98-106.
[3] Rytter JV, Helmark S, Chen J, et al. Synthetic promoter libraries forCorynebacteriumglutamicum[J].Applied Microbiology and Biotechnology, 2014, Published online.
[4] Alper H, Fischer C, Nevoigt E, et al. Tuning genetic control through promoter engineering[J].Proceedings of the National Academy of Sciences of the United States of America, 2005, 102(36):12678-12683.
[5] Cox RS 3rd, Surette MG, Elowitz MB. Programming gene expression with combinatorial promoters[J]. Molecular Systems Biology, 2007, 3:145-156.
[6] Murphy KF, Balazsi G, Collins JJ. Combinatorial promoter design for engineering noisy gene expression[J].Proceedings of the National Academy of Sciences of the United States of America, 2007, 104(31):12726-12731.
[7] Ellis T, Wang X, Collins JJ. Diversity-based, model-guided construction of synthetic gene networks with predicted functions[J]. Nature Biotechnology, 2009, 27(5):465-471.
[8] Nevoigt E, Fischer C, Mucha O, et al.Engineering promoter regulation[J].Biotechnology and Bioengineering, 2007, 96(3):550-558.
[9] Drummond DA, Iverson BL, Georgiou G, et al. Why high-error-rate random mutagenesis libraries are enriched in functional and improved proteins[J].Journal of Molecular Biology, 2005, 350(4):806-816.
[10] Kunichika K, Hashimoto Y, Imoto T. Robustness of hen lysozyme monitored by random mutations[J]. Protein Engineering, 2002, 15(10):805-809.
[11] Qin X, Qian J, Xiao C, et al. Reliable high-throughput approach for screening of engineered constitutive promoters in the yeastPichia pastoris[J].Letters in Applied Microbiology, 2011, 52(6):634-641.
[12] Klein-Marcuschamer D, Stephanopoulos G. Assessing the potential of mutational strategies to elicit new phenotypes in industrial strains[J].Proceedings of the National Academy of Sciences of the United States of America, 2008, 105(7):2319-2324.
[13] Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis[J].PCR Methods & Applications, 1992, 2(1):28-33.
[14] Leung DW, Chen E, Goeddel DV. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction[J].Technique, 1989, 1:11-15.
[15] Chusacultanachai S, Yuthavong Y. Random mutagenesis strategies for construction of large and diverse clone libraries of mutated DNA fragments[J]. Methods in Molecular Biology, 2004, 270:319-334.
[16] Wilson DS, Keefe AD. Random mutagenesis by PCR. Current Protocols in Molecular Biology, 2001, 51:8.3:8.3.1-8.3.9.
[17] Balci H, Ozturk MT, Pijning T, et al. Improved activity and pH stability ofE. coliATCC 11105 penicillin acylase by error-prone PCR[J].Applied Microbiology and Biotechnology, 2014, 98(10):4467-4477.
[18] Akbari M, Hansen MD, Halgunset J, et al. Low copy number DNA template can render polymerase chain reaction error prone in a sequence-dependent manner[J]. The Journal of Molecular Diagnostics, 2005, 7(1):36-39.
[19] Trollope KM, Nieuwoudt HH, Görgens JF, et al. Screening a random mutagenesis library of a fungal beta-fructofuranosidase using FT-MIR ATR spectroscopy and multivariate analysis[J].Applied Microbiology and Biotechnology, 2014, 98(9):4063-4073.
[20] McCullum EO, Williams BA, Zhang J, et al. Random mutagenesis by error-prone PCR[J].Methods Mol Biol, 2010, 634:103-109.
[21] Shafikhani S, Siegel RA, Ferrari E, et al. Generation of large libraries of random mutants inBacillus subtilisby PCR-based plasmid multimerization[J].Biotechniques, 1997, 23(2):304-310.
(责任编辑 李楠)
High-error-rate Random Mutagenesis of GAP Promoter in Pichia pastoris Using an Optimited Error Prone PCR
Qin Xiulin1Qian Jiangchao2Chu Ju2
(1. College of Life Science and Technology,Guangxi University,Nanning 530004;2. State Key Laboratory of Bioreactor Engineering,East China University of Science & Technology,Shanghai 200237)
The first important step toward a successful preparation of large and diverse promoter library with desired complexity, is to select a suitable mutagenesis strategy. To generate a promoter library of GAP promoter(pGAP)variants, mutations were introduced using error-prone PCR. After optimization of the conditions for EP-PCR random mutagenes, high mutation(error rate 1.1%)frequence was obtained using 1 ng/μL template and 10 mmol/L Mg2+, in combination with 25 thermal cycles. To increase mutational diversity and reach an appropriate error rate, three consecutive rounds of EP-PCR were carried out under the same conditions. After random sequencing of 10 clones from each round, an overall range of mutation rates from 1.1% to 4.0% was observed. Then, 250 clones containing pGAP variants were screened using the highthroughput screening approach in 48-deep-well plates. Among them, 5 mutants exhibited higher fluorescent intensity compared to the wildtype promoter.
Error-prone PCR Mutation frequency Random mutagenesis Pichia pastoris GAP promoter
2014-02-22
广西自然科学基金项目(2013GXNSFBA019096)
秦秀林,女,讲师,博士,研究方向:生物化学与分子生物学;E-mail:xiulinqin@gxu.edu.cn
钱江潮,女,教授,博士,研究方向:生物化学与分子生物学;E-mail:jiangchaoqian@ecust.edu.cn
