未培养技术在瘤胃产甲烷菌群研究中的应用
2014-03-17王兴文王加启赵圣国李发弟卜登攀
王兴文王加启赵圣国李发弟卜登攀
(1. 中国农业科学院北京畜牧兽医研究所 动物营养学国家重点实验室,北京 100193;2. 甘肃农业大学动物科学技术学院,兰州 730070)
未培养技术在瘤胃产甲烷菌群研究中的应用
王兴文1,2王加启1赵圣国1李发弟2卜登攀1
(1. 中国农业科学院北京畜牧兽医研究所 动物营养学国家重点实验室,北京 100193;2. 甘肃农业大学动物科学技术学院,兰州 730070)
瘤胃中的产甲烷菌为严格厌氧微生物,很难通过分离培养的方法进行研究。现已经培养并公布的瘤胃产甲烷菌只有4种,成千上万种类的瘤胃产甲烷菌不能培养出来。未培养技术的发展与应用突破了传统分离培养检测方法的限制,使瘤胃产甲烷菌的研究有了较大的进展。综述了实时定量PCR、16S rDNA文库、DGGE和高通量测序技术等6项具有代表性的未培养技术及其在瘤胃产甲烷菌研究中的应用,为以后瘤胃产甲烷菌的研究提供方法上的参考。
瘤胃 产甲烷菌 未培养技术 全基因组测序 高通量测序
反刍动物瘤胃中甲烷的生成会造成饲料能量的损失,同时甲烷也是造成温室效应的主要气体之一。它对温室效应的作用是CO2的25倍,全球每年由反刍动物排放的甲烷达8.1×107-9.2×107t,这相当于人类制造的甲烷的23%-27%[1]。产甲烷菌不足瘤胃微生物总量的4%,但几乎产生了反刍动物释放的全部甲烷[2]。将产甲烷菌的活性和数量控制在最适水平,达到反刍动物最佳的生产性能而对环境不造成影响是目前研究的重要方向。产甲烷菌是严格厌氧的古菌,一些在体内可以发现的产甲烷菌在体外很难分离培养。所以到目前为止,只有很少种类的产甲烷菌从瘤胃中分离出来,已经分离培养出来的瘤胃产甲烷菌主要为甲酸甲烷杆菌(Methanobacterium formicicum)、布氏甲烷杆菌(Methanobacterium byantii)、甲烷短杆菌(Methanobrevibacter ruminantium)和可移动的甲烷微菌(Methanomicrobium mobile)[3]。随着分子生物学的发展,未培养技术已越来越多地应用到瘤胃产甲烷菌的研究中,通过未培养技术可以不需要产甲烷菌的分离培养就可以准确地研究瘤胃中的产甲烷菌性质。本文主要介绍几种未培养技术以及其在产甲烷菌研究上的应用。
1 产甲烷菌多样性
1.1 产甲烷菌的引物
产甲烷菌研究中目的基因主要为16S rDNA和mcrA基因,16S rDNA是编码核糖体RNA(rRNA)分子的对应的DNA序列,既具有保守性,又具有高变性。保守性能够反应出生物物种的亲缘关系,为系统发育重建提供线索;高变性则能揭示出生物物种的特征核酸序列。mcrA基因编码甲基辅酶M还原酶(MCR)的α亚基,甲基辅酶M还原酶是甲烷产生过程中的一种关键酶,这种酶将与它的连接的甲基还原为CH4[4],它是产甲烷菌所独有的目的基因。16S rDNA的常见引物已在表1中列出,mcrA基因的常见引物已在表2中列出。
1.2 变性梯度凝胶电泳(Denaturing gradient gel electrophoresis,DGGE)
DGGE技术以DNA指纹图谱为基础,利用了DNA分子从双螺旋型到局部变性时电泳迁移率会急剧下降的原理。在得到的图谱中每一个条带都代表着某个微生物优势菌群。
Zhou等[26]以16S rDNA为目的基因利用DGGE研究两种不同日粮:生长期的日粮(高能量)和育肥期的日粮(低能量)对瘤胃产甲烷菌的影响。从DGGE的图谱中发现,两种日粮的优势条带不同,测序发现低能量日粮主要以Methanobrevibacter ruminantiumNT7为主,而高精料日料含有Methanobrevibacter smithii、Methanobrevibactersp. AbM4和M.ruminantiumNT7。Popova等[34]用高精料日粮和低精料日粮饲喂奶牛,以mcrA基因作为目的基因用DGGE的方法研究产甲烷菌的多样性,发现高精料日粮饲喂的奶牛产甲烷菌的多样性明显低于高粗料日粮组,日粮中的细胞壁成分的含量与瘤胃内的产甲烷菌的多样性成正比。Monica等[35]利用DGGE观察驯鹿两个时期(即驯鹿采食量的高峰期和低谷期)瘤胃产甲烷菌多样性发现,两组没有较大差别,说明采食量不会引起驯鹿瘤胃微生物的多样性的变化。
Zhou等[36]在奶牛饲粮中添加外源纤维素酶,甲烷生成量增加,为了解外源纤维分解酶对瘤胃产甲烷菌多样性和数量的影响以及甲烷产量和产甲烷菌群落结构的关系,应用实时定量PCR和DGGE技术,发现瘤胃产甲烷菌的数量没有改变,但是瘤胃产甲烷菌的多样性发生了改变。Cheng等[37]用3对引物(344fGC/915r、519f/915rGC和1106fGC/1378r)作DGGE研究中国西部绵羊产甲烷菌的多样性发现,引物519f/915rGC产生的条带分辨率最高。
DGGE直接利用微生物样品中的DNA或RNA对微生物群体加以区别,能解决微生物无法进行体外培养的难题,可判断出微生物间的种属关系,甚至还可以发现新的微生物。但是DGGE技术也存在一些缺点,如DNA的分离技术不同,细胞壁的破裂程度不同,DNA的回收率不同以及PCR不均等扩增都会造成试验结果的不同。此外,当微生物的DNA量不到样品DNA量的1%时,很难检测其条带。有一些产甲烷菌的基因组中含有多个目的基因的拷贝,就会过高的估计产甲烷菌的数量。
1.3 16S rDNA/mcrA基因建库
构建16S rDNA/mcrA文库可以对某一特定环境中甲烷菌群落结构和多样性进行分析。用PCR技术扩增特异的产甲烷菌的基因,然后将这种特异的基因与载体连接,转入大肠杆菌形成文库,选取阳性克隆测序,在测序的结果中去除载体和嵌合体序列,得到的序列与GenBank/RDP数据库比对,就可以知道每个克隆中的片段属于那一种微生物。
Franzolin等[38]研究不同粗饲料类型(牧草、玉米秸秆和甘蔗)对瘤胃产甲烷菌的影响,经16S rDNA建库后,得到467个克隆,包含19个操作分类单元:其中有4个在3个文库中都存在,8个是特异的,6个存在于玉米秸秆组和牧草组,1个存在于甘蔗组和牧草组,由此发现不同粗饲料类型会改变瘤胃产甲烷菌的多样性。Huang等[39]为了解牦牛甲烷产量少于青藏高原牛的原因,构建牦牛和青藏高原牛瘤胃产甲烷菌16S rDNA文库,在牦牛的文库中发现了209个克隆,青藏高原牛文库中205个克隆,将相似度在98%及以上归为一个操作分类单元,共发现了95个操作分类单元:牦牛单独含有46个操作分类单元,青藏高原牛含有34个操作分类单元,在两个文库中都存在的操作分类单元有15个。牦牛瘤胃的产甲烷多样性要高于青藏高原牛。

表1 产甲烷菌16S rDNA引物[5]

表2 产甲烷菌mcrA基因引物[5]
裴彩霞等[40]采用3对特异性引物扩增瘤胃古菌16S rRNA基因分别建立克隆文库研究晋南牛瘤胃产甲烷菌的多样性,引物Archf364/r1386建立的克隆库中,克隆分为4类,分别与Methanobrebacter. ap.1Y(61%)、SM9(23%)、NT7(14%)和AK-87(2%)相似。引物1Af/1100Ar建立的克隆库中,克隆分为两类,分别与Methanobacterium aarhusense(72%)和Methanosphaera stadtmanaeDSM3091(28%)相似。引物Met86F/Met1340R建立的克隆文库反映的古菌种类较为全面,除4种甲烷短杆菌(所占比例分别为47%、26%、11%和3%)外,还有Methanomicrobium mobile(2%)、以及类似Methanobacterium aarhusense(1%)和Methanosphaera stadtmanae(3%)的序列,还有7%的未匹配序列。Tymensen等[41]分别利用16S rDNA和mcrA建立文库研究瘤胃液和附着在原虫上的产甲烷菌的多样性,结果表明瘤胃液中产甲烷菌的多样性要显著高于附着在原虫上的产甲烷菌。Hook等[42]通过16S rDNA建库和DGGE的方法研究莫能菌素对瘤胃产甲烷菌多样性的影响发现,莫能菌素不会影响产甲烷菌的多样性。
建立基因文库较为准确,可以精确地了解瘤胃内产甲烷菌的系统分类。在实际研究中常常与DGGE相结合。但是要得到全面准确的数据,就要得到足够的克隆,挑取克隆的过程和步骤较为繁琐,测序的费用也相当高。而且即使有相当量的克隆也有可能遗漏一些丰度较低的基因。
1.4 限制性片段长度多态性(Restriction fragment length polymorphism,RFLP)技术
RFLP(T-RFLP)技术通过检测DNA在限制性内切酶酶切后形成的特定DNA片段的大小以识别限制性片段长度的多态性。现在已经发展出末端限制性片段长度多态性技术等,T-RFLP在技术上与RFLP相似,只是其中的一个引物的5'端用荧光物质标记,将PCR产物进行特异的酶切后就产生了许多不同长度的限制性片段,其中带有荧光标记的目标片段就可以被DNA自动测序仪检测到。
Frey等[43]为了了解泌乳牛瘤胃、十二指肠、回肠和粪便中产甲烷菌的多样性,应用RFLP技术研究发现在4个部位的产甲烷菌多样性差异不显著。Danielsson等[44]应用T-RFLP技术和16S rDNA建库结合研究不同日粮精粗比例对瘤胃产甲烷菌的影响,发现日粮精粗比与瘤胃产甲烷菌的多样性成反比。
1.5 高通量测序
高通量测序是通过对小亚基核糖体(如16S rRNA)基因高变区进行测序快速的检测样本中产甲烷菌的多样性。在转录组学研究中,RNA-seq直接定量检测瘤胃产甲烷菌的转录出来的mRNA的数量,从而了解产甲烷菌中代谢所需酶的活性[45,46]。
Fout等[47]利用高通量测序来检测瘤胃产甲烷菌多样性,得到138个序列,97%相似度序列归类分析,发现了20个OTU。其中18个OTU为数据库中已存在的序列,2个OTU为新菌,分别与Methanobrevibacter和Thermogymnomonas最为相似。
Hristov等[48]利用高通量测序检测日粮中添加不饱和脂肪酸对瘤胃中产甲烷菌的影响,发现添加不饱和脂肪酸可以明显增加Methanosphaera的数量而减少Methanobrevibacter的数量。
Lee等[49]研究韩国本地1种羊和3种牛的瘤胃产甲烷菌群落结构,高通量测序发现在4种动物的体内都不存在未知的产甲烷菌,都可以在数据库中找到。
2 产甲烷菌的定量分析
实时定量PCR(Real-time quantitative PCR,RTPCR)是在PCR的体系中加入荧光标记物后,模板扩增产物的量与荧光信号的强弱成正比,即每个模板的CT值与该模板的起始拷贝数的对数存在线性关系。利用已知拷贝数的标准品可做出标准曲线,只要得到未知样品的CT值,就能算出起始拷贝数。
Zhou等[25]以16S rDNA为目的基因用3种不同的特异性引物AbM4-F/AbM4-R,Stad-F/Stad-R和uniMet1-F/uniMet1-R分别定量测定Methanobrevibactersp. strain AbM4,M. stadtmanae和Total methanogens来研究日粮消化效率对牛瘤胃产甲烷菌数量的影响,发现消化率对总产甲烷菌的数量没有影响,但是高消化率组的Methanosphaera stadtmanae和Methanobrevibactersp. strain AbM4的数量是低消化组的1.92和2.26倍。Tymensen等[50]利用引物NestmetF/ NestmetR,Nest-MbbF/NestMbbR,NestRCCF/NestRCCR和NestMmF/NestMmR分别扩增总甲烷菌Methanobrevibacterspp.,RCC和Methanomicrobiumspp.来研究与原虫共生的产甲烷菌,发现Methanobrevibacterspp.和RCC是与原虫连接的主要产甲烷菌。Hook等[51]以mcrA基因为目的基因应用实时定量PCR和建库研究能量高低不同的日粮对奶牛瘤胃内的产甲烷菌的影响发现,日粮能量的高低对瘤胃产甲烷菌的影响不大。Denman等[30]用mcrA基因作实时定量PCR研究溴氯甲烷对瘤胃产甲烷菌的影响发现,日粮中添加溴氯甲烷可显著减少瘤胃中产甲烷菌的数量。David等[52]利用mcrA基因定量总产甲烷菌发现饲喂干草的绵羊瘤胃内的产甲烷菌要多于饲喂混合日粮的产甲烷菌。
实时定量PCR技术能够准确地定量研究瘤胃中产甲烷菌,但是要研究每一种产甲烷菌的数量必须得到相应的引物,但因一些产甲烷菌的引物还没有得到或在PCR过程中效率太低或特异性不好,因此不能定量研究。
3 产甲烷菌基因组
全基因组测序是对未知基因组序列的物种进行个体的基因组测序,从整体上研究微生物的功能和组成。在新西兰、澳大利亚和韩国正在进行着瘤胃产甲烷菌的基因组测序(表3)。
到目前为止,已有2种产甲烷菌测序完成并公布,第一个为Methanobrevibacter ruminantiumM1[53],它是由Bryant在1965年分离得到的,全长2.93 Mb。通过全基因组测序在M1的基因组中发现前噬菌体的基因组、分解酶基因以及内异肽酶PeiR基因,这些基因与M1的代谢息息相关。此外,还在M1的基因组中还发现了非核糖体肽合成酶基因,这是此基因在古菌基因组中第一次发现,从而揭示一些未知的代谢机理。另一个测序完成的产甲烷菌为Methanobrevibactersp. JHI,它是在韩国土牛瘤胃中分离得到的[54],通过基因组测序发现JHI的基因组与Methanobrevibactersp. AbM4(从新西兰羊皱胃分离,并测序完成,未发表)相似,说明它们在系统发育学上属于同一种类。但是在JHI的基因组中发现了前噬菌体基因,而AbM4没有,说明两者在进化上出现了分支。全基因组测序在JHI的基因组中也发现了与甲烷生成利用的H2、CO2和甲酸盐相关的酶。
通过全基因组测序可以清楚地了解目的产甲烷菌的代谢过程和系统发育的位置。在此基础之上,应用免疫方法和代谢阻遏物可以控制代谢过程中酶的活性来减少甲烷的排放[55]。但是全基因组测序是一项很大的工程,而且只有分离培养出来的产甲烷菌才能进行全基因组测序,成功分离培养的产甲烷菌只占瘤胃产甲烷菌很小的一部分。
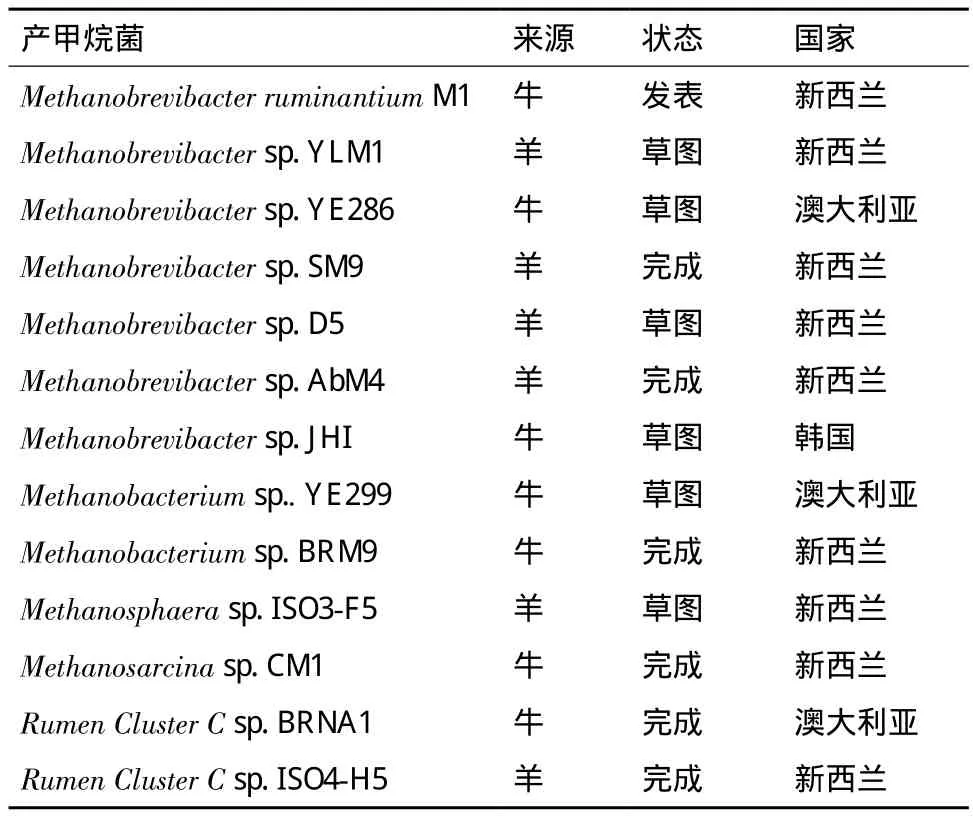
表3 瘤胃产甲烷菌基因组测序计划[56]
在实际的研究过程中,DGGE技术、16S rDNA/
mcrA基因建库、RFLP技术以及高通量测序主要用于研究产甲烷菌群落结构,包括菌落的多样性,丰富度。实时定量PCR主要用于定量产甲烷菌。若要了解产甲烷菌的功能和性质则需要全基因组测序。有时,为了更加准确具体地研究产甲烷菌,几项技术可以同时运用,克服彼此的缺点。如在一些研究中DGGE与16S rDNA/mcrA基因建库联合使用,DGGE可以发现主要的产甲烷菌,而16S rDNA/mcrA基因建库可以提高覆盖率全面地研究菌种的多样性,两者相互补充,互相验证。16S rDNA/mcrA基因建库与定量PCR的联合使用常常用于研究影响产甲烷菌的因素。
4 展望
未培养技术已经应用到了很多瘤胃产甲烷菌的研究中。随着高通量测序的普及,基于高通量测序的多样性研究是未来的发展趋势,全基因组测序也是未来研究产甲烷菌代谢途径和系统发育的重要内容。利用这些技术可以了解瘤胃中产甲烷菌的群落结构和生理状态,为合理有效控制甲烷的生成提供理论依据。
[1] IPCC(Intergovernmental Panel on Climate Change), Climate Change 2007[M]. The Scientific Basis. Cambridge, UK:Cambridge University Press, 2007.
[2] Lin C, Raskin L, Stahl DA. Microbial community structure in gastrointestinal tracts of domestic animals:comparative analyses using rRNA-targeted oligonucleotide probes[J]. FEMS Microbiol Ecol, 1997, 22:281-294.
[3] Janssen PH, Kirs M. Structure of the archaeal community of the rumen[J]. Appl Environ Microbiol, 2008, 74(12):3619-3625.
[4] Ellermann J, Hedderich R, Böcher R, et al. The final step in methane formation. Investigations with highly purified methyl-CoM reductase(component C)fromMethanobacterium thermoautotrophicum(strain Marburg)[J]. Eur J Biochem, 1988, 172(3):669-677.
[5] Zhou M, McAllister TA, Guan LL. Molecular identification of rumen methanogens:Technologies, advances and prospects[J]. Anim Feed Sci Tech, 2011, 166-167:76-86.
[6] Yanagita K, Kamagata Y, Kawaharasaki M, et al. Phylogenetic analysis of methanogens in sheep rumen ecosystem and detection ofMethanomicrobiummobile By fluorescencein situhybridization[J]. Biosci Biotechnol Biochem, 2000, 64(8):1737-1742.
[7] Whitford MF, Teather RM, Forster RJ. Phylogenetic analysis of methanogens from the bovine rumen[J]. BMC Microbiol, 2001, 1:5.
[8] Skillman LC, Evans PN, Naylor GE, et al. 16S ribosomal DNA-directed PCR primers for ruminal methanogens and identification of methanogens colonising young lambs[J]. Anaerobe, 2004, 10(5):277-285.
[9] Tajima K, Nagamine T, Matsui H, et al. Phylogenetic analysis of archaeal 16S rRNA libraries from the rumen suggests the existence of a novel group of archaea not associated with known methanogens[J]. FEMS Microbiol Lett, 2001, 200(1):67-72.
[10] Wright AD, Williams AJ, Winder B, et al. Molecular diversity of rumen methanogens from sheep in Western Australia[J]. Appl Environ Microbiol, 2004, 70(3):1263-1270.
[11] Shin EC, Choi BR, Lim WJ, et al. Phylogenetic analysis of archaea in three fractions of cow rumen based on the 16S rDNA sequence[J]. Anaerobe, 2004, 10(6):313-319.
[12] Whitehead TR, Cotta MA. Phylogenetic diversity of methanogenic archaea in swine waste storage pits[J]. FEMS Microbiol Lett, 1999, 179(2):223-226.
[13] DeLong EF. Archaea in coastal marine environments[J]. Proc Natl Acad Sci USA, 1992, 89:5685-5689.
[14] Tokura M, Chagan I, Ushida K, et al. Phylogenetic study of methanogens associated with rumen ciliates[J]. Curr Microbiol, 1999, 39(3):123-128.
[15] Marchesi JR, Weightman AJ, Cragg BA, et al. Methanogen and bacterial diversity and distribution in deep gas hydrate sediments from the Cascadia Margin as revealed by 16S rRNA molecular analysis[J]. FEMS Microbiol Ecol, 2001, 34(3):221-228.
[16] Embley TM, Finlay BJ, Thomas RH, et al. The use of rRNA sequences and fluorescent probes to investigate the phylogenetic positions of the anaerobic ciliateMetopus palaeformisand its archaeobacterial endosymbiont[J]. J Gen Microbiol, 1992, 138(7):1479-1487.
[17] Barns SM, Fundyga RE, Jeffries MW, et al. Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment[J]. Proc Natl Acad Sci USA, 1994, 91(5):1609-1613.
[18] Baker GC, Smith JJ, Cowan DA. Review and re-analysis of domainspecific 16S primers[J]. J Microbiol Methods, 2003, 55(3):541-555.
[19] Yu Z, García-González R, Schanbacher FL, et al. Evaluations of different hypervariable regions of archaeal 16S rRNA genes in profiling of methanogens by Archaea-specific PCR and denaturing gradient gel electrophoresis[J]. Appl Environ Microbiol, 2008, 74(3):889-893.
[20] Bano N, Ruffin S, Ransom B, et al. Phylogenetic composition of Arctic Ocean archaeal assemblages and comparison with Antarctic assemblages[J]. Appl Environ Microbiol, 2004, 70(2):781-789.
[21] Pinar G, Saiz-Jimenez C, Schabereiter-Gurtner C, et al. Archaeal communities in two disparate deteriorated ancient wall paintings:detection, identification and temporal monitoring by denaturing gradient gel electrophoresis[J]. FEMS Microbiol Ecol, 2001, 37:45-54.
[22] Kleikemper J, Pombo SA, Schroth MH, et al. Activity and diversity of methanogens in a petroleum hydrocarbon-contaminated aquifer[J]. Appl Environ Microbiol, 2005, 71(1):149-158.
[23] Wright AD, Pimm C. Improved strategy for presumptive identification of methanogens using 16S riboprinting[J]. J Microbiol Methods, 2003, 55(2):337-349.
[24] van Hoek AH, van Alen TA, Sprakel VS, et al. Multiple acquisition of methanogenic archaeal symbionts by anaerobic ciliates[J]. Mol Biol Evol, 2000, 17(2):251-258.
[25] Amann RI, Ludwig W, Schleifer KH. Phylogenetic identification andin situdetection of individual microbial cells without cultivation[J]. Microbiol Rev, 1995, 59(1):143-169.
[26] Zhou M, Hernandez-Sanabria E, Guan LL. Assessment of the microbial ecology of ruminal methanogens in cattle with different feed efficiencies[J]. Appl Environ Microbiol, 2009, 75(20):6524-6533.
[27] Hales BA, Edwards C, Ritchie DA, et al. Isolation and identification of methanogen-specific DNA from blanket bog peat by PCR amplification and sequence analysis[J]. Appl Environ Microbiol, 1996, 62(2):668-675.
[28] Luton PE, Wayne JM, Sharp RJ, et al. The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill[J]. Microbiology, 2002, 148(11):3521-3530.
[29] Hallam SJ, Girguis PR, Preston CM, et al. Identification of methyl coenzyme M reductase A(mcrA)genes associated with methaneoxidizing archaea[J]. Appl Environ Microbiol, 2003, 69(9):5483-5491.
[30] Denman SE, Tomkins NW, McSweeney CS. Quantitation and diversity analysis of ruminal methanogenic populations in response to the antimethanogenic compound bromochloromethane[J]. FEMS Microbiol Ecol, 2007, 62(3):313-322.
[31] Scanlan PD, Shanahan F, Marchesi JR. Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis[J]. BMC Microbiol, 2008, 8:79.
[32] Steinberg LM, Regan JM. Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge[J]. Appl Environ Microbiol, 2008, 74(21):6663-6671.
[33] Juottonen H, Galand PE, Yrjälä K. Detection of methanogenic Archaea in peat:comparison of PCR primers targeting the mcrA gene[J]. Res Microbiol, 2006, 157(10):914-921.
[34] Popova M, Martin C, Eugène M, et al. Effect of fibre and starchrich finishing diets on methanogenicArchaeadiversity and activity in the rumen of feedlot bull[J]. Anim Feed Sci Tech, 2011, 166-167:113-121.
[35] Sundset MA, Edwards JE, Cheng YF, et al. Rumen microbial diversity in Svalbard reindeer, with particular emphasis on methanogenic archaea[J]. FEMS Microbiol Ecol, 2009, 70(3):553-562.
[36] Zhou M, Chung YH, Beauchemin KA, et al. Relationship between rumen methanogens and methane production in dairy cows fed diets supplemented with a feed enzyme additive[J]. J Appl Microbiol, 2011, 111(5):1148-1158.
[37] Cheng YF, Mao SY, Liu JX, et al. Molecular diversity analysis of rumen methanogenic Archaea from goat in eastern China by DGGE methods using different primer pairs[J]. Lett Appl Microbiol, 2009, 48(5):585-592.
[38] Franzolin R, St-Pierre B, Northwood K, et al. Analysis of rumen methanogen diversity in water buffaloes(Bubalus bubalis)under three different diets[J]. Microb Ecol, 2012, 64(1):131-139.
[39] Huang XD, Tan HY, Long R, et al. Comparison of methanogen diversity of yak(Bos grunniens)and cattle(Bos taurus)from
the Qinghai-Tibetan plateau, China[J]. BMC Microbiol, 2012, 12:237.
[40] 裴彩霞, 毛胜勇, 朱伟云. 晋南牛瘤胃中古菌分子多样性的研究[J]. 微生物学报, 2008, 48(1):8-14.
[41] Tymensen LD, Beauchemin KA, McAllister TA. Structures of freeliving and protozoa-associated methanogen communities in the bovine rumen differ according to comparative analysis of 16S rRNA and mcrA genes[J]. Microbiology, 2012, 158(7):1808-1817.[42] Hook SE, Northwood KS, Wright AD, et al. Long-term monensin supplementation does not significantly affect the quantity or diversity of methanogens in the rumen of the lactating dairy cow[J]. Appl Environ Microbiol, 2009, 75(2):374-380.
[43] Frey JC, Pell AN, Berthiaume R, et al. Comparative studies of microbial populations in the rumen, duodenum, ileum and faeces of lactating dairy cows[J]. J Appl Microbiol, 2010, 108(6):1982-1993.
[44] Danielsson R, Schnürer A, Arthurson V, et al. Methanogenic population and CH4production in swedish dairy cows fed different levels of forage[J]. Appl Environ Microbiol, 2012, 78(17):6172-6179.
[45] Zened A, Combes S, Cauquil L, et al. Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets[J]. FEMS Microbiol Ecol, 2013, 83(2):504-514.
[46] Thoetkiattikul H, Mhuantong W, Laothanachareon T, et al. Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16S rRNA gene pyrosequencing[J]. Curr Microbiol, 2013, 67(2):130-137.
[47] Fouts DE, Szpakowski S, Purushe J, et al. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen[J]. PLoS One, 2012, 7(11):48289.
[48] Hristov AN, Callaway TR, Lee C, et al. Rumen bacterial, archaeal, and fungal diversity of dairy cows in response to ingestion of lauric or myristic acid[J]. J Anim Sci, 2012, 90(12):4449-4457.
[49] Lee HJ, Jung JY, Oh YK, et al. Comparative survey of rumen microbial communities and metabolites across one caprine and three bovine groups, using bar-coded pyrosequencing and1H nuclear magnetic resonance spectroscopy[J]. Appl Environ Microbiol, 2012, 78(17):5983-5993.
[50] Tymensen L, Barkley C, McAllister TA. Relative diversity and community structure analysis of rumen protozoa according to T-RFLP and microscopic methods[J]. J Microbiol Methods, 2012, 88(1):1-6.
[51] Hook SE, Steele MA, Northwood KS, et al. Impact of high-concentrate feeding and low ruminal pH on methanogens and protozoa in the rumen of dairy cows[J]. Microb Ecol, 2011, 62(1):94-105.
[52] Yáñez-Ruiz DR, Macías B, Pinloche E, et al. The persistence of bacterial and methanogenic archaeal communities residing in the rumen of young lambs[J]. FEMS Microbiol Ecol, 2010, 72(2):272-278.
[53] Leahy SC, Kelly WJ, Altermann E, et al. The genome sequence of the rumen methanogenMethanobrevibacter ruminantiumreveals new possibilities for controlling ruminant methane emissions[J]. PLoS One, 2010, 5(1):8926.
[54] Lee JH, Rhee MS, Kumar S, et al. Genome sequence ofMethanobrevibactersp. strain jh1, isolated from rumen of Korean native cattle[J]. Genome Announc, 2013, 1(1):2-13.
[55] Wedlock DN, Janssen PH, Leahy SC, et al. Progress in the development of vaccines against rumen methanogens[J]. Animal, 2013, 7(2):244-252.
[56] Morgavi DP, Kelly WJ, Janssen PH, et al. Rumen microbial(meta)genomics and its application to ruminant production[J]. Animal, 2013, 7(1):184-201.
(责任编辑 狄艳红)
The Application of Uncultured Methods in the Study of Ruminal Methanogen Population
Wang Xingwen1,2Wang Jiaqi1Zhao Shengguo1Li Fadi2Bu Dengpan1
(1. State Key Laboratory of Animal Science,Institute of Animal Science,Chinese Academy of Agricultural Science,Beijing 100193;2. College of Animal Science and Technology,Gansu Agricultural University,Lanzhou 730070)
The ruminal methanogen are strictly anaerobic, which are hard to culture.The ruminal methanogen which have been cultured successfully are only 4 species.while hundreds of species of methanogen could not be cultured.The application and development of uncultured technology promotes the research of ruminal methanogen, which overcome the limitation of the tranditional technology of isolation and culturing. Several dominant culture-independent technologies used to study ruminal methanogens were introduced in this paper to provide
on methods for future ruminal methanogen study.
Rumen Methanogen Uncultured methods Whole genome sequencing High throughput DNA sequencing
2013-09-03
国家自然科学基金项目(31261140365),科技支撑计划项目(2012BAD12B02),中国科学院知识创新工程重要方向项目(XDA01010301)
王兴文,男,硕士研究生,研究方向:反刍动物营养;E-mail:xing.wen111@163.com
卜登攀,副研究员,硕士生导师,研究方向:反刍动物营养与代谢调控;E-mail:burdengpan@gmail.com
