环三磷腈基六臂氯化聚醚粘合剂的合成与热性能①
2014-03-15卢先明苏海鹏葛忠学
甘 宁,肖 啸,刘 庆,卢先明,苏海鹏,葛忠学
(西安近代化学研究所,西安 710065)
0 引言
聚氨酯是固体推进剂包覆用的一种衬层基体材料,通常是借助于自由装填式装药完成对药柱包覆的。操作时,将由端羟预聚物、异氰酸酯固化剂和无机(或有机)填料组成的液体基料浇铸到盛有推进剂药柱的模具中,通过粘合剂的羟基与固化剂的异氰酸酯基之间的氨酯化反应,将推进剂药柱与衬层粘接在一起。聚氨酯衬层的特点是粘接强度高、低温力学性能优良[1-3],但其不足之处在于由于分子结构的限制,耐热性能不太理想,只能作为内孔燃烧固体推进剂的衬层,不能用于端面燃烧固体推进剂的衬层。为解决聚氨酯材料耐热性能差的缺陷,甘孝贤等[4]以具有碳氮六元杂环基本骨架的三羟乙基异氰脲酸酯为引发剂,环氧氯丙烷为单体,三氟化硼乙醚络合物为催化剂,按照阳离子开环聚合反应机理合成出主链上具有碳氮杂环结构的液体氯化聚醚预聚物,该预聚物既具有刚性的碳氮六元杂环型骨架,又具有阻燃元素氯及较高的(N+O)/C原子个数比,因而赋予材料较高的耐热性、耐烧蚀性。它与异氰酸酯化合物相匹配,就可完成固化。目前,该材料已按照贴壁浇注工艺用于某型号推进剂的绝热包覆[5]。
然而,遗憾的是目前没有看到国内外有关含磷氮杂环结构氯化聚醚多元醇的合成与应用方面的报道。因此,本研究设想以耐热、耐烧蚀性更佳的磷氮六元杂环结构(即环三磷腈)取代上述碳氮六元杂环杂环,为固体推进剂绝热包覆合成出一种耐热、耐烧蚀性更好的环三磷腈基六臂氯化聚醚粘合剂。
1 实验
1.1 试剂与仪器
六氯环三磷腈,自制,熔点,113 ℃;对羟基苯甲醛,CR,ALDRICH,使用前用水重结晶后真空干燥;苯酚钠,AR,成都市科龙化工试剂厂;四氢呋喃,AR,成都市科龙化工试剂厂,使用前脱水处理;硼氢化钠,AR,成都市科龙化工试剂厂;甲醇,AR,成都市科龙化工试剂厂;环氧氯丙烷(ECH),分析纯,成都市科龙化工试剂厂,使用前4Å分子筛干燥;1,2-二氯甲烷,分析纯,成都市科龙化工试剂厂,使用前4Å分子筛干燥;甲苯,AR,成都市科龙化工试剂厂,使用前4 Å分子筛干燥;三氟化硼乙醚络合物(BF3·Et2O),分析纯,天津市百世化工有限公司;六(4-醛基苯氧基)环三磷腈(PN-6CHO),自制[6],熔点161~162 ℃;六(4-羟甲基苯氧基)环三磷腈(PN-6OH),自制[6],熔点220~221 ℃。
KQ-250DB型数控超声波反应器,昆山市超声仪器有限公司;红外光谱采用美国Nicolet公司的Nexus 870型傅里叶变换红外光谱仪测定;核磁共振谱采用瑞士Bruker AV 500型核磁共振仪测定;动态热失重分析(TGA)用Universal V2.6D TA Instruments测定,升温速率为10 ℃/min,测试范围为室温至800 ℃,气氛为空气;相对平均分子质量采用蒸汽渗透压法(VPO)测定。
1.2 合成路线
HCPCP的合成路线如下所示:以六(4-羟甲基苯氧基)环三磷腈(PN-6OH)为多元醇引发剂,三氟化硼乙醚络合物为催化剂,引发环氧氯丙烷阳离子开环聚合得到目标聚合物HCPCP。


1.3 环三磷腈基六臂氯化聚醚HCPCP的合成
室温下,向配置机械搅拌、温度计、滴液漏斗、回流冷凝管和超声水浴的250 ml四口烧瓶中依次加入100 ml的1,2-二氯乙烷和100 ml甲苯,在搅拌下缓慢加入4.37 g(5 mmol)PN-6OH,超声下搅拌30 min。然后将反应体系加热至40~50 ℃,一次性加入3.8 ml(30 mmol)BF3·Et2O,超声下搅拌15 min,然后缓慢滴加8.33 g(90 mmol)环氧氯丙烷,控制滴加速度使反应体系温度保持40~50℃;待反应体系中的固体全部消失后停止滴加环氧氯丙烷,待反应体系温度降至0~5 ℃后继续滴加剩余环氧氯丙烷,加毕后,0 ℃恒温反应24 h。反应完成后加水终止反应,经后处理,得到外观透明的淡黄色粘性液体。


图1 HCPCP的1H NMR
2 结果与讨论
2.1 HCPCP的合成机理
视催化剂用量与单体引入方式的不同,环单体的阳离子开环聚合机理分为活性链端机理(ACE)和活性单体机理(AMM)[7]。当催化剂/引发剂的摩尔比≤1,且单体缓慢滴加时,聚合反应倾向于AMM机理。依据AMM机理能够对相对分子质量及官能度进行有效控制。当催化剂/引发剂的摩尔比>1,且单体滴加较快时,聚合反应倾向于ACE机理,此时活性链端易发生分子内及分子间的链转移反应,形成低相对平均分子质量的齐聚物或冠醚,直接影响聚合物的相对分子质量与官能度。本研究所用的引发剂六(4-羟甲基苯氧基)环三磷腈具有6个活性反应位点,为了能更好控制聚合物的相对分子质量及官能度,必须使聚合反应更倾向于活性单体机理。因此,需严格控制催化剂用量和单体的滴加速率。

图2 HCPCP的13C NMR谱

对应氢编号abcδ6.74~6.947.07~7.112.05~2.07氢原子数112对应氢编号d+efgδ3.71~3.783.66~3.691.14~1.16氢原子数321

表2 HCPCP的13C NMR谱化学位移与对应结构
2.2 聚合反应介质的选择
由于PN-6OH的熔点较高,且为固相凝聚态,故无法与环氧单体进行本体聚合,只能从均相溶液与非均相溶液聚合方法入手。PN-6OH仅能溶解于二甲基甲酰胺、二甲基亚砜、丙酮、四氢呋喃等强极性溶剂中。以二甲基亚砜为溶剂,进行均相聚合时发现,无论在高温还是低温,均得不到聚合物。二甲基甲酰胺则较为特殊,它先与催化剂BF3·Et2O在低温条件下形成稳定的络合物,得不到活性中心,无法进行聚合反应。而当温度升高到80 ℃时,二甲基甲酰胺与BF3·Et2O形成的络合物迅速分解,释放出活性中心,是开环聚合反应猛烈进行,反应失去控制,得不到理想的聚合物。丙酮、四氢呋喃由于其结构中含有活性很高的氧原子,会与催化剂BF3·Et2O迅速形成络合物,聚合反应也无法进行。
实验发现,以二氧六环、环己酮、甲苯、二氯甲烷、1,2-二氯乙烷等惰性溶剂为分散剂,聚合反应则能够平稳进行,预聚物能够很好地溶解于反应介质中。如果以二氧六环、环己酮为溶剂,它们可参与开环聚合,反应无法控制;二氯甲烷熔点较低,聚合反应时放热量较大,易使二氯甲烷挥发,影响反应进行。因此,只能选择1,2-二氯乙烷和甲苯作为反应介质。实验中发现:(1)无论是1,2-二氯乙烷还是甲苯,都不能溶解引发剂PN-6OH,但聚合反应却都能有效地进行。不同的是固体引发剂进入液相的速率不同,甲苯体系较快,反应液粘度较大,且未参与反应的部分形成质地坚硬的大结块;而1,2-二氯乙烷体系较慢,反应液粘度较小,且未参与反应的部分形成松散体。(2)无论如何变化反应条件,2种反应体系始终有20%~30%的引发剂没有参与反应。究其原委,乃是由于:(1)甲苯有利于增大链引发速率,但对预聚物溶解性差;(2)1,2-二氯乙烷有利于链的增长,且对预聚物溶解性好;(3)非完全进入液相的粘性反应中间体将未反应的高熔点引发剂包裹形成结块,造成引发剂与环单体隔绝,中断聚合。这一切都说明溶剂的溶解度参数直接主宰着PN-6OH的消耗速率。将2种溶剂按一定比例混合后,却取得了很好的效果。本研究以反应体系的固体残余量(即反应剩余固体占引发剂投料量的比例)为考察对象,研究了不同溶剂体系对聚合反应的影响,具体结果见表3。

表3 甲苯/1,2-二氯乙烷体积比对引发剂残余量的影响
由表3数据可发现,随着1,2-二氯乙烷比例的增大,固体残余量明显减少,当甲苯/1,2-二氯乙烷=1/3时,固体结块完全消失;继续增加1,2-二氯乙烷的比例,固体残余量反而增大。说明混合溶剂由于溶解度参数的调节,使得聚合体系既有较高的链引发速率,又对初级聚合物有较高的溶解能力。因此,将混合溶剂的体积比确定为甲苯/1,2-二氯乙烷=1/3。
2.3 催化剂用量
催化剂用量是影响阳离子开环聚合反应及聚合物品质的关键因素之一。本研究根据阳离子开环聚合反应的特点,并结合以往经验,对催化剂用量对聚合反应的影响进行了详细考察,见表4。表4中,OH/ECH摩尔比为1/3,Mn=2 538;收率以经后处理所得聚合物质量与理论产量的比值表示;引发温度40~50 ℃,聚合温度0~5 ℃,聚合反应时间为24 h。
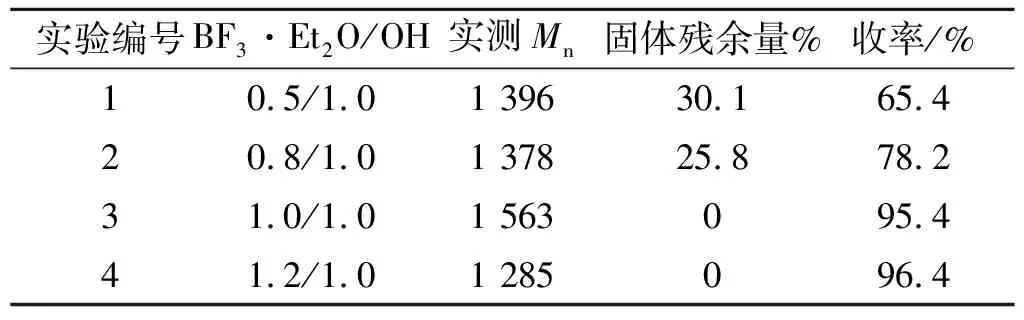
表4 催化剂用量对反应的影响
由表2可知,当羟基与催化剂等摩尔比时所得聚合物的相对平均分子质量最大,反应收率最高;当催化剂用量过少时,实测聚合物相对平均分子质量与理论相对平均分子质量相差较大,且反应收率较低。这是因为引发剂PN-6OH具有6个活性羟基引发点,催化剂用量过少时,6个活性引发点不能同时进行链引发,造成部分引发点链增长很快,非完全引发的预聚物将未反应的起始剂包裹起来,从而阻断了聚合反应;当催化剂用量过高时,由于反应倾向于活性链端机理,聚合反应过程中容易出现分子间的链转移反应(导致聚合物相对平均分子质量分布较宽)和分子内的链“回咬”反应(导致冠醚和小相对平均分子质量齐聚物的生成),从而使聚合反应中的冠醚与不含有环三磷腈结构的齐聚醚等副产物生成量大大增加,一方面导致环状单体消耗过多而阻碍链增长反应,直接造成聚合物相对平均分子质量较低;另一方面,聚合反应过程中产生的冠醚及小分子齐聚物在后处理过程中均被除去,导致反应收率大大降低。
2.4 单体滴加时间
阳离子聚合具有快引发、快增长、易转移、难终止等特点,这决定了聚合无需较长时间;活性可控聚合机理要求聚合时溶液中的自由单体浓度应尽可能的低,这就对单体的滴加速度提出了较为严格的要求,为此进行了较为详细的研究,结果见表5。表5中,OH/ECH摩尔比为1/3,Mn=2 538;收率以经后处理所得聚合物质量与理论产量的比值表示;引发温度:40~50 ℃,BF3·Et2O/OH/ECH摩尔比为1/1/3,聚合温度0~5 ℃,聚合反应时间为24 h。

表5 单体滴加时间对反应的影响
分析表5可得出如下结论:当单体滴加时间小于4 h时,聚合物的产率及相对平均分子质量均较低。原因在于过快的滴加速率导致反应体系中的单体量相对“过剩”,而这些“过剩”的单体容易发生副反应,使聚合反应倾向于ACE机理,此时活性链端易发生分子内及分子间的链转移反应,生成大量的低相对平均分子质量齐聚物或冠醚(其对相对平均分子质量和收率的影响同2.3节);当滴加时间为4 h时,聚合物的相对平均分子质量最高,效果最好;再进一步延长滴加时间,相对平均分子质量和产率变化不大。原因在于环氧氯丙烷活性较高,在一定的滴加速度下(滴加4 h左右)能够保证瞬时上链聚合。
2.5 HCPCP后处理
环氧氯丙烷在进行阳离子开环聚合时副反应较多,易形成冠醚和无环三磷腈结构的齐聚醚,严重时可占总量的20%~30%。由于和环三磷腈预聚醚在结构上的差异,这种冠醚和齐聚物与环三磷腈预聚物相溶性较差,混合在一起造成外观上不透明。为了获得高品质的HCPCP,必须对粗产品进行精制,将那些不含环三磷腈结构的齐聚醚除去。
将异丙醇/石油醚按1∶1体积比配成萃取剂。氯化聚醚HCPCP /萃取剂按1/1体积比投料,搅拌回流0.5 h,停止搅拌,自行降温,使体系分相,待降至20~50℃,用虹吸法抽出上层萃取液,下层溶液浓缩得到精制预聚物。如此重复萃取2次,取样测定相对平均分子质量,评价萃取效果,结果见表6。表6中,萃取前Mn=1 550,萃取前样品不透明;收率以经后处理所得聚合物质量与理论产量的比值表示。
表6中数据表明,用混合溶剂萃取可获得较为满意的结果。当萃取温度低于30 ℃时,萃取前后相对平均分子质量相差不大,且产物外观仍稍带浑浊,说明萃取温度低将会影响萃取效果;当萃取温度高于30 ℃时,所得聚合物Mn在1 600以上且外观透明,说明30 ℃以上的萃取温度均能将冠醚和齐聚醚萃取干净。此外,由表6可看出,随萃取温度升高,反应收率会出现明显的下降趋势。这是因为高萃取温度会造成聚合物在萃取剂中溶解度增大,部分低相对平均分子质量的聚合物在萃取过程中随冠醚和齐聚物一起被除去,而造成目标产物的损失。

表6 萃取温度对反应的影响
2.6 HCPCP的化学结构
由表4~表6数据可明显看出,无论怎样变化反应条件,甚至经过萃取处理,相对平均分子质量的实测值与理论值相差较大,预聚物HCPCP的Mn在1 500~1 600之间波动,说明采用PN-6OH这种特殊的化合物作为引发剂时,只能使环氧氯丙烷进行链的引发,难以发生链的增长。换言之,大多数情况下每个苄醇只能与1个环氧氯丙烷反应(理论Mn=1 428),过量的环氧氯丙烷形成无环三磷腈结构的聚醚副产物。
采用如下方法证明了PN-6OH上的6个羟基已全部引发了环氧氯丙烷:将HCPCP在氢氧化钠作用下进行端部关环反应,可得到以环三磷腈为内核的多臂环氧树脂(HCPEP),其理想结构如下所示:

对关环产物进行红外光谱分析,发现红外光谱中3 421 cm-1处苄醇特有的吸收峰消失,而在812 cm-1的处出现了中等强度的环氧基团特征吸收峰,说明6个羟基全部参与反应。HCPCP及HCPEP的红外图谱如图3所示。

图3 HCPCP与HCPEP的红外图谱对比
综上所述,PN-6OH中的6个羟基作为活性点全部引发ECH聚合,聚合产物HCPCP确为以环三磷腈为内核的六臂氯化聚醚结构。此外,根据引发剂PN-6OH(Mn=873)的结构和HCPCP的Mn在1 500~1 600范围内的事实,说明引发剂分子上的每个苄醇在大多数情况下只能引发1个环氧氯丙烷上链,即只发生链引发反应。据此可认为,HCPCP应具有如下结构:

当然,这种结构对于本研究是最理想的结果,因为每个氯原子所处的位置正好在每个臂的链端,而且每个臂上只有1个氯原子。从材料的力学性能和耐热性角度讲,六臂结构能够使材料成型后具有较大的交联密度,保证材料较高的强度,而最短的脂肪链能够使材料保持优良的耐热性。
3 HCPCP的热性能
3.1 玻璃化转变温度
图4为Mn=1 600的HCPCP的玻璃化转变温度曲线,并与线性环氧氯丙烷开环聚合物(PECH,Mn=2 000~2 500)进行了比较。
由图4可知,HCPCP的Tg=-37.98 ℃,而线型PECH的Tg=-38~-39 ℃,二者基本相当。通常情况下,向聚醚主链上引入刚性杂环结构,会降低分子链的柔顺性,从而使玻璃化温度Tg升高。然而,本研究所合成的HCPCP并没有表现出明显的Tg升高现象,这可能是因为支化或超支化高分子与线型高分子链的结构存在着很大差异。Nielsen L E等[8-9]用数值计算的方法,研究了超支化高分子玻璃化温度与体系端基和支化点密度的关系。据此分析,与线型PECH相比,本研究合成的HCPCP的自由体积增大,端基密度相对减小,同时分子链的活动受到的约束程度不断减小,从而使玻璃化温度相对降低。

图4 HCPCP和PECH的Tg曲线
3.2 热稳定性
通过测试样品的TGA曲线,研究了HCPCP在高温条件下的热稳定性。样品的热分析测试条件:空气环境,从30 ℃升至800 ℃,升温速率10 ℃/min。图5为HCPCP的TGA和DTG曲线。
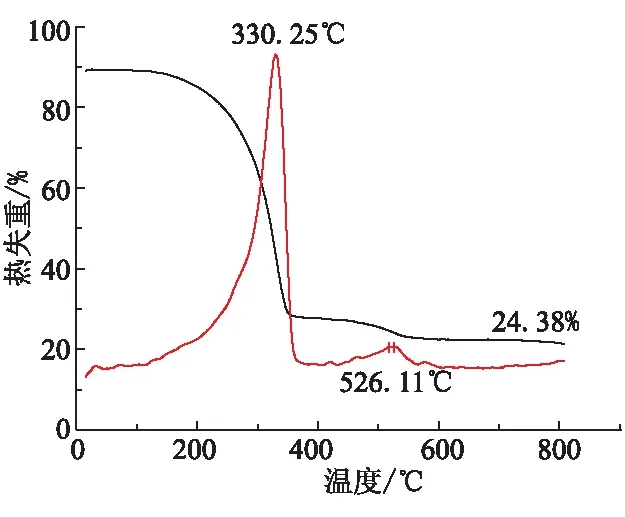
图5 HCPCP的TGA和DTG曲线
HCPCP的热分解主要分为2个阶段:在280~330 ℃之间失重约75%,可认为是聚合物侧基的分解及聚醚主链的无规降解;在526 ℃时发生第二次热分解,这可能是环三磷腈结构的磷氮单双键发生断裂,生成了线型磷氮链状分子片段;当温度达到600 ℃以上时,不再发生明显的热分解和热失重现象,这是因为线型磷氮分子片段相互之间发生交联反应,生成磷氮氧网状交联结构;当温度升高到800 ℃时,残焦量为24.38%。
4 结论
(1)以六氯环三磷腈、对羟基苯甲醛和环氧氯丙烷为原料,依次经亲核取代反应、还原反应和阳离子开环聚合反应合成出以环三磷腈为内核的六臂氯化聚醚HCPCP,并确定了合成HCPCP的最佳反应条件。
(2)热性能研究表明,HCPCP的玻璃化转变温度为-37.98 ℃,与线性环氧氯丙烷开环聚合物的玻璃化温度基本相当,表明HCPCP的低温柔顺性较好;HCPCP在800 ℃时的残焦量为24.38%,而线性环氧氯丙烷开环聚合物在800 ℃时已经完全分解,残焦量几乎为零,说明环三磷腈结构的引入,有助于提高氯化聚醚多元醇的耐热性。
参考文献:
[1] 张世约,甘孝贤,刘凤云.碳氮杂环型氯化聚醚多元醇的合成研究[J].火炸药学报,1993,1:6-8.
[2] 甘孝贤,张世约,何颖,等.碳氮杂环多异氰酸酯固化剂的合成研究[J].火炸药学报,1993,2:11-13.
[3] 赵凤起.国外无(少)烟聚氨酯包覆层研制情况[J].火炸药学报,1993,16(1):15.
[4] 甘孝贤.碳氮杂环型氯化聚醚多元醇的合成研究[J].火炸药,1993,1:6-8.
[5] 甘孝贤,等.推进剂包覆层用聚氨酯粘合剂[P].ZL03105889.2,2006.
[6] Gabino A C,Lucia F C.On the synthesis of functionalized cyclic and polymeric aryloxyphosphazenes from phenols[J].Journal of Applied Polymer Science,1996,59:1879-1885.
[7] Takeo S,Yoshihiko H,Shu-ichi M.Ring-opening polymerization of oxycyclobutane by boron trifluoride.Concentration of propagating species and rate of propagation[J].Macromolecules,1971,4:1-3.
[8] Nielsen L E.Cross-linking effect on physical properties of polymers[J].J.Macromol.Sci.Rev.Macromol.Chem.,1969,3(1):69-103.
[9] Stutz H,Mertes J.A generalized theory for the glass transition temperature of crosslinked and uncrosslinked polymers[J].J.Polym.Sci.:Part B:Polym.Phys.,1990,28(9):1483-1498.
