口蹄疫病毒实时定量RT-PCR方法的建立及初步应用
2014-03-08何于雯信爱国李华春
胡 骑,何于雯,信爱国,李华春
(云南省畜牧兽医科学院云南省热带亚热带动物病毒重点实验室,云南昆明 650224)
口蹄疫(Food and mouth disease,FMD)是由小核糖核酸病毒科(Picornaviridae)口蹄疫病毒属(Aphthovirus)的FMD 病毒(FMDV)引起的偶蹄动物传染病。FMDV 存在7 种相互无交叉保护性免疫应答的不同血清型(O、A、C、SAT-1、SAT-2、SAT-3 和Asia1)[1-2]。FMDV 能够在BHK-21、IB-RS-2、羔羊或牛睾丸细胞等多种细胞上复制。通常采用观察FMDV 细胞病变效应(CPE)和测定病毒组织半数感染量(TCID50)的方法来分析FMDV 的复制效率。随着具有快速、敏感、特异性强的实时定量RT-PCR(qRT-PCR)技术在FMDV 基因表达水平的分析、病原体的定性和定量检测等方面得到了广泛的应用[3-6],通过建立qRT-PCR 方法分析FMDV 在细胞培养物中的复制动态,测定FMDV 即时增殖滴度,明确病毒的复制效率,是对上述传统方法的有益补充。
本实验针对FMDV 3D 基因,建立检测FMDV的SYBR Green I qRT-PCR 方法,以BHK-21 细胞18S rRNA 为内参基因,采用双标准曲线法[7]分析FMDV 感染细胞总RNA 中FMDV 基因组当量(Genome equivalents per μg total RNA,GE/μg),应用该方法对亚洲1 型FMDV 兔化弱毒ZB(att)株和其体外拯救病毒vZB 株感染BHK-21 细胞后的病毒复制动态进行了测定,同时测定病毒一步生长曲线,进行比较分析。该方法为FMDV 的检测和研究FMDV 增殖规律及致病性等相关研究提供了一种快速评价方法。
1 材料和方法
1.1 病毒株和细胞系 FMDV 亚洲1 型兔化弱毒ZB 株细胞适应毒ZB(att)(DQ533483)、ZB(att)株感染性克隆pZBatt 体外拯救病毒vZB[8]以及用于FMDV qRT-PCR 特异性验证的蓝舌病病毒(BTV)、猪瘟病毒(CSFV)和猪繁殖障碍综合征病毒(PPRSV)由云南省畜牧兽医科学院保存。BHK-21 传代细胞系由中国农业科学院哈尔滨兽医研究所于力研究员惠赠。
1.2 主要试剂 病毒总RNA 提取试剂TRIpure Reagent 购自北京艾德莱生物科技有限公司;cDNA合成试剂盒和荧光定量PCR 试剂盒购自北京全式金生物技术有限公司;2×Master Mix Taq 酶、DNA MarkerⅠ均购自北京艾德莱生物科技有限公司。
1.3 引物设计 采用文献报道的位于FMDV 基因组高度保守区RNA 聚合酶基因(3D)内的引物序列[9]和根据真核生物18S rRNA 序列(NR003278)设计引物,经NCBI Blast 比对验证,建立检测FMDV 的qRT-PCR 方法。采用文献报道的检测亚洲1 型FMDV VP1 基因的上、下游引物As1DF 和P33 引物进行RT-PCR[10]。引物序列见表1。引物由上海生工生物工程技术服务股份有限公司合成。
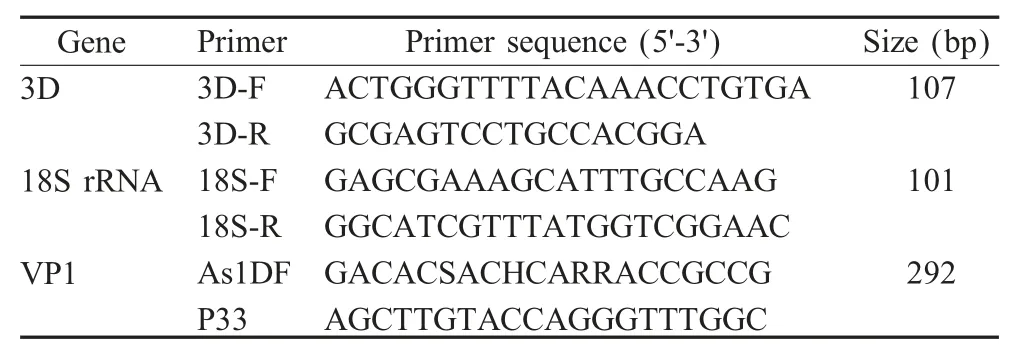
表1 RT-PCR 和qRT-PCR 引物Table 1 Primers of RT-PCR and qRT-PCR
1.4 病毒RNA的提取和c DNA的合成 将拯救病毒vZB(10-4.5TCID50/0.1 mL)200 μL,按RNA 提取试剂说明书方法提取基因组RNA,采用oligo dT18为反转录引物,合成cDNA 用于RT-PCR 和qRTPCR 试验。
1.5 qRT-PCR方法的建立 以合成的cDNA 为模板,分别对FMDV 3D 基因和内参18S rRNA 基因进行扩增,建立检测FMDV 的实时荧光定量PCR。反应总体积为20 μL,其中2×TranStart Green qPCR Mix 10 μL,上、下游引物(10 μM)各0.5 μL,模板cDNA 1 μL,ROX Reference Dye II 0.5 μL。反应条件:95 ℃10 s,95 ℃5 s,60 ℃30 s 收集荧光,共扩增45 个循环;最后添加溶解曲线生成步骤。同时设立阴性对照和空白对照。
采用双标准曲线法对细胞悬液中的FMDV RNA进行相对定量,检测标准品为质粒pT-FMDV-3D(稀释梯度为10-2~10-7),18S rRNA 标准品为质粒pT-18r(稀释梯度为10-2~10-7)。将两者进行梯度稀释,分别绘制标准曲线。各实验组样品的Ct 值与标准曲线对比,计算检测样品的浓度。经内参基因18S rRNA归一化处理后,计算细胞悬液总RNA 的FMDV F值(GE/μg)。归一化值=目的基因浓度/内参基因浓度,F 值=(待测组样品目的基因浓度/待测组样品内参基因浓度)/(对照组样品目的基因浓度/对照组样品内参基因浓度)。结果采用Excel 和Graphpad prism 5 软件进行统计分析和绘图。
1.6 qRT-PCR方法敏感性、特异性及重复性试验10 倍倍比稀释vZB(10-4.5TCID50/0.1 mL),取200 μL提取总RNA,进行SYBR Green I qRT-PCR 试验,确定病毒检测的最低稀释度,同时以检测亚洲1 型FMDV VP1 基因的上、下游引物As1DF 和P33 进行常规的RT-PCR 扩增[10],比较分析该方法的敏感性。提取BTV、CSFV、PRRSV 的RNA,进行qRT-PCR检测,验证其特异性。将同一批次的标准品质粒模板和阳性样品ZB(att)进行梯度稀释,提取RNA,采用建立的qRT-PCR 检测,每个浓度重复3 次,隔天再重复3 次,求平均值、标准差和变异系数。
1.7 样品的检测 将拯救病毒vZB(10-4.5TCID50/0.1 mL)和亲本病毒ZB(att)(10-4.83TCID50/0.1 mL)稀释成100×TCID50/0.1 mL,各取0.2 mL 分别接种12孔板中长满单层BHK-21 细胞,每个稀释度接种3孔。在接种后第4 h,8 h,12 h,16 h 和24 h 分别收获病毒,测定其TCID50,绘制一步生长曲线;同时利用所建立的qRT-PCR 方法检测同一时间收获病毒的RNA 相对含量,经3 次重复试验取其几何平均数进行分析,比较验证两种方法的符合率。
2 结果
2.1 标准质粒的构建 采用oligo dT18 为反转录引物,以vZB 培养病毒悬液提取总RNA 为模板,合成cDNA。采用引物18S-F 和18S-R 扩增细胞内参对照基因,采用引物3D-F 和3D-R 扩增FMDV 高度保守区3D 基因,分别获得101 bp 和107 bp PCR 产物(图1)。将上述PCR 产物分别克隆至pMD18-T中,获得重组质粒pT-FMDV-3D 和pT-18S 作为检测FMDV 基因组的定量质粒标准品。

图1 FMDV 3D 基因片段和细胞18S rRNA 片段的PCR 扩增产物Fig.1 Amplification of FMDV 3D gene and 18S rRNA fragments by RT-PCR
2.2 标准曲线的建立 分别将pT-FMDV-3D(383 ng/μL)和pT-18S(388 ng/μL)10 倍倍比稀释,各取6 个不同稀释度(10-2~10-7)作为模板进行荧光定量PCR扩增,生成扩增动力曲线和标准曲线(图2)。FMDV 3D 基因的标准曲线方程为:Ct=-3.61×log(conc)+4.21,熔解温度为83.6 ℃;18S rRNA 标准曲线方程为:Ct=-3.54×log(conc)+1.19,熔解温度为80.7 ℃。

图2 SYBR Green I qRT-PCR 检测FMDV-3D 和18S rRNA 的标准曲线Fig.2 Standard curve of the qRT-PCR for FMDV-3D and 18S rRNA
2.3 敏感性试验 将10 倍倍比稀释的病毒vZB(10-4.5TCID50/0.1 mL),各取200 μL,提取RNA,反转录后进行qRT-PCR 检测,结果显示能够最低检测到101.5×TCID50/0.1 mL 样品,倍比稀释在100~10-3呈现较好的线性关系,敏感性比常规的RT-PCR 高10 倍,其敏感性试验的扩增动力曲线及常规RT-PCR 检测见图3。
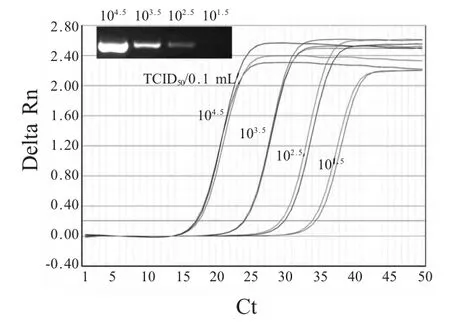
图3 不同稀释度FMDV vZB 株的qRT-PCR 检测和常规RT-PCR 检测结果Fig.3 The detection of FMDV vZB strain by the qRT-PCR and traditional RT-PCR at different dilutions
2.4 特异性试验 提取BTV、CSFV 和PRRSV 的RNA,利用建立的检测FMDV 的qRT-PCR 进行特异性检测,同时设无模板对照。结果表明,仅FMDV 阳性标准品出现阳性信号,其他病毒样品和未加模板对照的检测结果均为阴性(图略)。
2.5 重复性试验 将标准质粒pT-FMDV-3D 和pT-18S 进行10 倍连续梯度稀释,经过预试验选择10-2~10-7作为浓度梯度,用相同的荧光定量PCR 反应体系和反应程序,分3 个试验批次做FMDV-3D和18S rRNA 基因的标准曲线,每个浓度梯度做3次重复。结果显示3 批阳性样品和阴性样品重复扩增3 次的变异系数均小于2.8 %,表明所建立的qRT-PCR 检测方法具有较高的可重复性(表2)。
表2 SYBR GreenⅠqRT-PCR 的组内和组间重复性试验结果Table 2 Intra-assay and inter-assay reproducibility test of the qRT-PCR
2.6 FMDV一步生长曲线 病毒一步生长曲线结果表明,vZB 和ZB(att)在BHK-21 细胞中的复制能力相近,4 h 测定病毒TCID50分别为10-2.00TCID50/0.1 mL和10-2.17TCID50/0.1 mL,病毒滴度达到峰值所需的时间均在16 h 以后,24 h CPE 均达100 %,vZB 和ZB(att)的毒价分别为10-5.17TCID50/0.1 mL 和10-5.33TCID50/0.1 mL(图4)。

图4 ZB(att)和vZB 的一步生长曲线Fig.4 One-step growth curves of the ZB(att)and vZB
2.7 qRT-PCR检测细胞总RNA中FMDV基因组当量 分别提取拯救病毒vZB 和亲本毒ZB(att)在接种BHK-21 细胞后不同时间点分别收获细胞的总RNA,对FMDV RNA 进行定量分析。结果显示ZB(att)和vZB 在4 h 测定细胞悬液内病毒RNA 水平分别为101.08GE/μg 和101.02GE/μg;至24 h,细胞均产生100 % CPE,病毒RNA 水平分别为102.81GE/μg和102.80GE/μg(图5)。同一时间点方差分析(p>0.05),差异不显著,两株病毒在BHK-21 细胞内复制效率相似。该结果与病毒一步生长曲线结果相比较,两种方法均表明vZB 和亲本毒ZB(att)在BHK-21 细胞中复制效率相似。
3 讨论
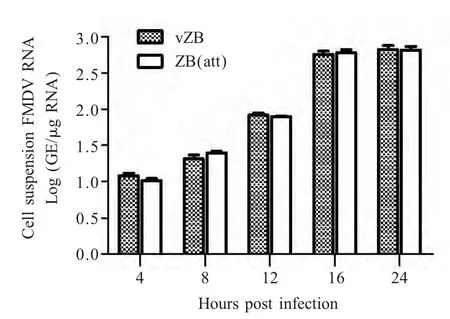
图5 接毒后不同时间细胞悬液内ZB(att)和vZB 病毒RNA 水平的变化Fig.5 Cell suspension ZB(att)and vZB RNA levels at different time points post infection
本研究以FMDV 细胞培养物中的18S rRNA 作为内参基因,建立了检测FMDV 的qRT-PCR 方法,用于细胞培养物中FMDV RNA 的相对定量分析。该方法的内参基因的选择,最初参考文献报道的GAPDH 为内参基因建立相对定量表达法测定FMDV 在体外复制[6],显示GAPDH 基因溶解曲线有小杂峰(图略),后改用18S rRNA 作为内参,获得了溶解曲线为单一峰的特异性扩增曲线。但分别以GAPDH 和18S rRNA 为内参基因,对同一处理的样品,重复测定3 次,分析FMDV RNA 水平,其结果是一致的。对实验数据分析,常规采用2-ΔΔCT法分析表达差异,要求内参基因和目标基因扩增效率一致。本实验采用双标准曲线法进行分析,虽然每次试验都分别用标准品做内参基因18S rRNA 和目的基因FMDV 3D 的标准曲线,并同时扩增各个样本中目的基因和内参基因,但该方法分析简单,实验条件容易优化,从各自标准曲线上可以直接测得样本中初始量,将目的基因表达量进行归一化处理以消除初始RNA 的差异带来的表达水平差异[7,11],先计算每个样本的归一化值,再计算细胞悬液总RNA的病毒基因组当量F 值,结果采用柱形图表示[12]。该方法特异性强,重复性好,比常规RT-PCR 灵敏10 倍。本实验初步应用该方法对亚洲1 型FMDV 兔化弱毒ZB(att)株和其基因工程拯救病毒vZB 株在BHK-21 细胞上的复制效率进行了测定和分析,其结果显示体外拯救病毒与其亲本病毒的复制动态相似,结果与TCID50测定的病毒一步生长曲线一致。这结果与王海伟等和李平花等建立的Asia 1 型FMDV 的感染性克隆分子拯救病毒与亲本病毒的毒力比较也基本一致[13-14]。该方法对FMDV 的快速检测和分析FMDV 突变株的病毒复制效率差异提供了一种快速评价方法。
[1]Grubman M J,Baxt B.Foot-and-mouth disease[J].Clin Microbiol Rev,2004,17(2):465-493.
[2]Mason P W,Grubman M J,Baxt B.Molecular basis of pathogenesis of FMDV[J].Virus Res,2003,91(1):9-32.
[3]Shaw A E,Reid S M,Ebert K,et al.Implementation of a one-step real-time RT-PCR protocol for diagnosis of foot-andmouth disease[J].J Virol Methods,2007,143(1):81-85.
[4]石立立,顾潮江,张倩,等.应用改良的实时Taq Man 荧光定量RT-PCR 技术检测口蹄疫病毒及其3D 基因转录水平[J].中国病毒学,2006,21(5):449-453.
[5]李永东,张常印,姜平,等.口蹄疫病毒实时RT-PCR 检测方法的建立[J].中国人兽共患病学报,2006,22(3):212-215.
[6]Du Jun-zheng,Gao Shan-dian,Luo Ji-huai,et al.Effective inhibition of foot-and-mouth disease virus(FMDV)replication in vitro by vector-delivered microRNAs targeting the 3D gene[J].Virol J,2011,8:292-295.
[7]唐永凯,贾永义.荧光定量PCR 数据处理方法的探讨[J].生物技术,2008,18(3):89-91.
[8]Xin Ai-guo,Li Hua-chun,Li Le,et al.Genome analysis and development of infectious cDNA clone of a virulence-attenuatedstrain of foot-and-mouth disease virus type Asia 1 from China[J].Vet Microbiol,2009,138(3-4):273-280.
[9]Callahan J D,Brown F,Osorio F A,et al.Use of a portable real-time reverse transcriptase-polymerase chain reaction assay for rapid detection of foot-and-mouth disease virus[J].J Am Vet Med Assoc,2002,220(11):1636-1642.
[10]Vangrysperre W,De Clercq K.Rapid and sensitive polymerase chain reaction based detection and typing of foot-and-mouth disease virus in clinical samples and cell culture isolates,combined with a simultaneous differentiation with other genomically and/or symptomatically related viruses[J].Arch Virol,1996,141(2):331-344.
[11]徐丽华,刘春雷,常玉梅,等.双标准曲线相对定量PCR试验原理与方法[J].生物技术通报,2011,196(1):70-75.
[12]黎刚.HCV 非结构蛋白和结构蛋白在复制和感染中的作用[D].上海:第二军医大学,2009.
[13]王海伟,涂亚斌,周国辉,等.Asia1 型口蹄疫病毒感染性克隆的建立[J].中国预防兽医学报,2008,30(12):915-919.
[14]李平花,白兴文,卢曾军,等.口蹄疫病毒Asia1/JS/China/2005 株基因组全长感染性克隆的构建[J].畜牧兽医学报,2009,40(5):706-711.
