DNA 条形码揭示日本纺织娘(Mecopoda niponensis)个体内和个体间的序列变异
2014-01-14周志军尚娜常岩林石福明
周志军 尚娜 常岩林 石福明
(1. 河北大学生命科学学院,保定 071002;2. 河北大学图书馆,保定 071002)
DNA 条形码技术是基于线粒体DNA 细胞色素C 氧化酶亚基I 基因(Mitochondrial COI)5'端一段约650 bp 的标准基因片段在DNA 水平上进行物种区分[1],已被广泛用于鱼类、鸟类、哺乳类、鳞翅目、鞘翅目、双翅目、甲壳类及洞穴蜘蛛等的物种鉴定[2-11]。在利用DNA 条形码进行物种鉴定过程中,关于划分物种的序列分歧的“阈值”问题一直存在较大的争议。Hebert 等[1]曾提出基于DNA 条形码序列分歧划分物种的“阈值”,无脊椎动物为3%,鸟类和哺乳类为2%。Yang 等[11]研究发现,北京百花山地区夜蛾科的种间平均遗传距离(11.29%)远大于种内(0.03%),采用1%作为种间序列差异阈值的鉴定准确率可达95%。Meyer 等[12]对维多利亚湖9 属14 种丽鱼科鱼研究发现,尽管这些物种间具有明显的形态学和生态学变异,但其序列分歧<1%。Hebert 等[6]基于对北美260 种鸟的研究再次提出相对种间分歧界定,即种间差异≥种内变异10 倍。但是,该物种界限缺乏生物学支持,在一些地理分布广泛或亲缘关系很近的物种间经常存在种间差异和种内变异重叠现象[13-15]。
线粒体假基因(Nuclear sequences of mitochondrial origin,numts)广泛存在于不同生物类群的基因组中[16-21],在使用线粒体COI 作为DNA 条形码进行物种鉴定时,核内与其同源的numts 极易被共扩增,导致物种鉴定错误和物种多样性高估。通常认为,由于核基因组中的numts 失去了其原有功能,进化方式不同于mtDNA 序列,无净化选择(purify selection),各种突变容易积累(终止密码子、插入/缺失及移码突变等),因此,多数numts 可以通过序列比对或在翻译成氨基酸后被识别出来并加以剔除[5],只有近期转移入核的numts 才难以被识别[22,23]。Moulton 等[24]研究发现,在直翅目昆虫中存在一些核内numts,由于不存在插入/缺失、提前出现终止密码子及移码突变等现象,而难以与线粒体COI 区分,并建议使用氨基酸序列进行核查并加以剔除,剔除标准为碱基替代导致≥2 个氨基酸非同义替换。
纺织娘亚科Mecopodinae 我国仅记录1 属2种,即纺织娘属Mecopoda Audinet-Sermille,1831的纺织娘M. elongata L.,1758 和日本纺织娘M. niponensis De Haan,1842[25],其作为鸣虫在我国具有悠久的人工饲养历史。两种纺织娘种间区别并不困难,但由于分布广泛,种内存在一定的形态变异。此前,国内外尚无关于这两种纺织娘之间关系的研究。GenBank 数据库中仅有我们提交的纺织娘(JQ917910)和日本纺织娘(JQ917909)线粒体基因组全序列[26]。为探讨DNA 条形码技术在其物种鉴定中的可行性,本研究对我国分布的这两种纺织娘标准DNA 条形码片段进行了测定,并对在日本纺织娘中发现的线粒体COI 极高的numts 和异质性(或无法使用常规方法甄别的numts)现象进行了分析,试图从DNA 条形码数据的后期质量控制着手,对numts 进行甄别,以保证基于DNA 条形码进行物种鉴定的准确性。
1 材料与方法
1.1 材料
本研究所用两种纺织娘标本信息如表1。野外采集到的标本,立即置于无水酒精中浸泡保存,次日更换酒精,带回实验室后于4℃冰箱中保存备用。
1.2 方法
1.2.1 基因组DNA 提取、PCR 扩增及测序 使用TIANamp Genomic DNA Kit(天根,北京),按照操作说明,取酒精浸泡标本的后足股节肌肉组织进行DNA 提取。根据GenBank 数据库中收录的直翅目线粒体基因组数据修改后的引物LCO1490(5'-TCAACAAATCATAAGGACATTGG-3')和HCO2198(5'-TAAACTTCAGGGTGTCCAAAGAATCA-3')被用于扩增DNA 条形码标准区段[27]。PCR 扩增反应体系为:总DNA 模 板 3 μL,DNA 聚 合 酶 2.5 U(TaKaRa),dNTPs 0.2 mmol/L,引物各1 mmol/L,超纯水补足50 μL。扩增条件为:94℃预变性4 min;94℃变性30 s,50℃退火30 s,70℃延伸90 s,共35 个循环;70℃延伸7 min。PCR 扩增产物经琼脂糖凝胶电泳切胶纯化后,送上海铂尚生物技术有限公司进行双向测序。最初PCR 扩增采用宝生物rTaq DNA 聚合酶进行,PCR 扩增产物经1%琼脂糖凝胶电泳切胶回收、纯化后,全部日本纺织娘个体直接测序结果峰图杂乱,表明可能存在多种扩增产物。因此,改用宝生物高保真LA Taq DNA 聚合酶重新进行PCR 扩增,使用PMD®-19T 载体(宝生物,大连)进行克隆,并随机挑取5-17 个单克隆测序。
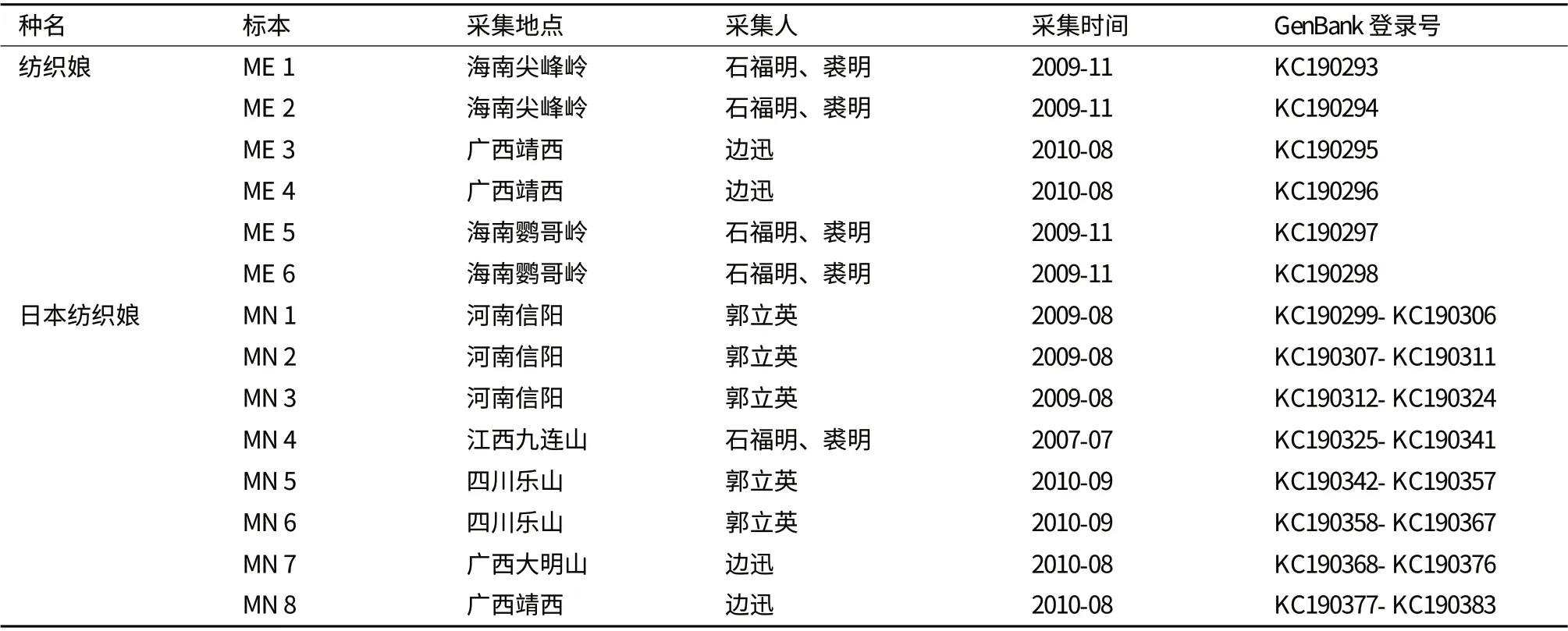
表1 样本信息
1.2.2 序列分析 测序结果导入Lasergene 6 软件包中的SeqMan 软件[28],进行序列拼接与手工校正,确定分析序列。为避免核基因组中numts 序列的干扰,拼接好的序列,首先使用Lasergene 6 中的EditSeq 软件[28]筛查存在终止密码子的numts,然后使用序列比对软件ClustalX[29]筛查存在碱基插入/缺失和移码突变的numts,最后使用MEGA 5.0[30]分析剩余序列对应的氨基酸序列变异位点,参考Moulton 等[24]标准,将碱基突变引起≥2 个氨基酸非同义替换作为numts 筛选标准,对近期由线粒体转入核内的numts 进行筛选。利用MEGA 5.0[30]计算两种纺织娘种间、种内个体间及日本纺织娘同一个体内部不同克隆之间的Kimura 2-parameter(K2P)遗传距离,并基于邻接法构建系统发育关系树。
2 结果
2.1 线粒体COI序列分析与numts确定
使用DNA 条形码通用引物LCO1490/HCO2198进行PCR 扩增,两种纺织娘均可获得单条清晰、整齐的靶标条带。6 个纺织娘个体的PCR 法直接测序结果良好,序列拼接比对发现靶标片段长度均为658 bp,无碱基插入/缺失、移码突变及翻译提前终止等现象,用无脊椎动物线粒体基因组密码子表推断产生的氨基酸序列完全一致。8 个日本纺织娘个体的PCR 法直接测序结果峰图杂乱,而克隆法测定的85 个克隆拼接后的序列长度介于580-662 bp。以此前基于L-PCR 技术结合嵌套PCR 二次扩增获得的线粒体基因组全序列中的COI 序列作为参照序列(JQ917909)[26],发现其中20 条克隆序列存在终止密码子或碱基插入/缺失,为明显的numts(4 条由于异常终止密码子出现致使翻译终止,15 条存在碱基插入/缺失,1 条存在读码框摆动);在剩余的65条长度均为658 bp,无终止密码子的克隆序列中发现38 条序列含≥2 个氨基酸非同义替换位点的疑似numts,氨基酸序列变异情况见图1。
2.2 遗传距离分析
K2P 模型分析显示,两种纺织娘之间的K2P 距离为0.077,纺织娘及日本纺织娘不同个体间的K2P距离分别为0-0.008 及0.007-0.044,个体内不同克隆之间的K2P 距离分别为0-0.143(含numts)及0-0.076(不含numts)(表2)。
2.3 系统发育树构建
利用邻接法(Neighbor-Joining,NJ)选择K2P遗传距离模型,同时采用1 000 次Bootstrap 重复抽样检验各分支置信度,构建系统发育树(图2)。由图2 可见,全部纺织娘个体以极高的置信度(99%)聚为一个单系分支G,而日本纺织娘的聚类结果比较复杂,5 条numts 序列在系统树基部形成两个分支H 和I,3 条numts 序列与代表纺织娘的分支G 形成姊妹分支F,分支B(仅1 条序列)和E 均为numts序列,分支D 由单条COI 序列形成,分支C 及A 则分别主要由numts 序列及COI 序列形成。

图1 日本纺织娘线粒体COI 基因氨基酸序列变异位点
3 讨论
许多学者将DNA 条形码区分物种失败的原因归咎于近期分化的物种间存在的不完全谱系分选现象。本研究通过对两种纺织娘14 个个体的线粒体COI序列进行拼接和校正后发现,日本纺织娘的线粒体COI 存在极高的numts 和异质性(或无法使用常规方法甄别的numts),这些序列聚类形成多个不同分支。造成此现象的原因可能有两种:(1)检测到的日本纺织娘线粒体COI 变异来自PCR 扩增错误。为了减少PCR 扩增错误所带来的影响,本研究采用了高保真的TaKaRa LA Taq 酶,如此高频率的突变不能简单归因于PCR 扩增错误。(2)日本纺织娘线粒体COI 本身即存在极高的numts 和异质性。类似现象在直翅目其他类群中已有报道。例如,Bensasson等[17]发现在红股秃蝗Podisma pedestris 中存在多条不同的线粒体COI-like 序列;Moulton 等[24]研究发现在Anabrus simplex 等直翅目昆虫中存在一些线粒体COI 序列,它们没有翻译提前终止、碱基插入/缺失或移码突变等,但其推导的氨基酸序列存在较多的非同义突变,认为其实际为numts 序列而非异质性。我们认为,虽然不能排除少量由于PCR 扩增错误所引起的变异,但绝大多数的日本纺织娘线粒体COI 变异是其自身即具有的numts 和异质性。遗憾的是,目前尚无方法具体区分PCR 扩增错误和真实的线粒体DNA 分子变异。
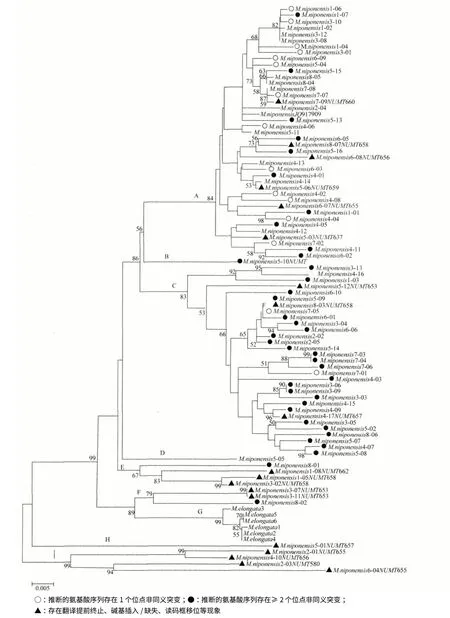
图2 基于Kimura 2-parameter 模型构建的两种纺织娘线粒体COI 和numts NJ 树

表2 基于Kimura 2-parameter 模型的日本纺织娘个体内不同克隆间和个体间遗传距离
参考Moulton 等[24]标准,以采用L-PCR 法获得的线粒体COI 序列为参照(JQ917909)[26],我们将≥2 个氨基酸非同义替换作为numts 的剔除标准,取得了一定效果,但聚类结果仍并不是十分理想,主要表现在:(1)在疑似numts 的分支C 中包含两条仅含一个非同义替换位点的序列;(2)由单条COI 序列形成的分支D 在NJ 树上的位置显示,其与主要由线粒体COI 序列组成的分支A 的距离较疑似numt 的分支C 远;(3)在主要由线粒体COI 序列组成的分支A 中包含数条存在≥氨基酸非同义替换的疑似numts 序列。究其原因,可能是由于日本纺织娘的进化过程中存在多次线粒体DNA 向核内转移的事件,导致大量没有翻译提前终止、插入/缺失和移码突变的numts 出现,即这些numts 由线粒体DNA 新近转移至核内。
Numts 共扩增现象对于使用DNA 条形码技术进行物种鉴定的干扰必须予以重视。为此,一些研究人员先后提出了多种高效甄别numts 的方法,如设计物种特异性引物进行扩增、L-PCR 扩增mtDNA 大片段作为模板进行二次扩增及反转录PCR等[24,31-34]。但是,DNA 条形码之所以备受推崇,并得以广泛使用,其优势就在于其无需预知所研究材料的遗传背景,且操作方法简单,易于标准化,而上述几种方法都不同程度地违背了这些DNA 条形码技术成立的基本条件。我们认为,在使用DNA 条形码技术进行物种鉴定的过程中,只能从数据的后期质量控制着手,对numts 进行甄别,例如,Chung和Steiper[35]使用HKY85 模型构建NJ 树,根据系统发育聚类结果对numts 进行甄别。在系统发育树上,numts 可能形成独立分支,但是,由于相似的进化速率及近期的mtDNA 核整合事件,numts 和真实的mtDNA 序列间分歧很小,用核苷酸序列可能很难区分。Numts 序列曾被认为是古线粒体DNA 向细胞核基因组转移的“分子化石”[36],而实际上,这种遗传物质的水平转移自真核生物出现以来就一直在持续进行。
4 结论
利用DNA 条形码通用引物对我国分布的两种纺织娘14 个个体进行了扩增与测定。6 个纺织娘个体均可用PCR 产物直接测序法获得良好测序结果,而8 个日本纺织娘个体测序结果峰图杂乱。改用克隆法测定的85 条克隆序列长度介于580-662 bp,其中,20 条为明显的numts(4 条存在终止密码子,15 条存在碱基插入/缺失,1 条存在读码框摆动),38 条为疑似numts(与参考序列相比含≥2 个非同义替换位点)。邻接法聚类结果显示,纺织娘聚类形成一个单系分支(自举值为99),而日本纺织娘的线粒体COI 和numts 序列则聚类形成多个分支,表明这些numts 序列可能是线粒体COI 多次向核内转移的结果。
[1] Hebert PDN, Cywinska A, Ball SL, et al. Biological identifications through DNA barcodes[J]. P Roy Soc B-Biol Sci, 2003, 270(1512):313-321.
[2] 常虹, 郝德君, 肖荣堂, 等. 基于线粒体COI 基因的齿小蠹属昆虫DNA 条形码研究[J]. 昆虫学报, 2012, 55(9):1075-1081.
[3] 程磊, 常玉梅, 鲁翠云, 等.鲫属鱼类DNA 条码及种与亚种划分[J]. 动物学研究, 2012, 33(5):463-472.
[4] Greenstone MH, Rowley DL, Heimbach U, et al. Barcoding generalist predators by polymerase chain reaction:carabids and spiders[J]. Mol Ecol, 2005, 14(10):3247-3266.
[5] Hebert PDN, Penton EH, Burns JM, et al. Ten species in one:DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator[J]. PNAS, 2004, 101(41):14812-14817.
[6] Hebert PDN, Stoeckle MY, Zemlak TS, et al. Identification of Birds through DNA Barcodes[J]. PloS Bio, 2004, 2(10):e312.
[7] Kerr KCR, Stoeckle MY, Dove CJ, et al. Comprehensive DNA barcode coverage of North American birds[J]. Mol Ecol Notes, 2007, 7(4):535-543.
[8] Kumar NP, Rajavel AR, Natarajan R, et al. DNA barcodes can distinguish species of Indian mosquitoes(Diptera :Culicidae)[J]. J Med Entomol, 2007, 44(1):1-7.
[9] 马英, 李海龙, 亮鲁, 等. DNA 条形码技术在青海海东地区小型兽类鉴定中的应用[J]. 生物多样性, 2012, 20(2):193-198.
[10] Smith MA, Wood DM, Janzen DH, et al. DNA barcodes affirm that 16 species of apparently generalist tropical parasitoid flies(Diptera, Tachinidae)are not all generalists[J]. PNAS, 2007, 104(12):4967-4972.
[11] 杨聪慧, 韩辉林, 迟美妍, 等. DNA 条形码技术在北京百花山地区夜蛾科物种鉴定中的应用[J]. 昆虫学报, 2012, 55(9):1082-1092.
[12] Meyer A, Kocher TD, Basasibwaki P, et al. Monophyletic origin of Lake Victoria cichlid fishes suggested by mitochondrial DNA sequences[J]. Nature, 1990, 347(6293):550-553.
[13] Moritz C, Cicero C. DNA barcoding:promise and pitfalls[J]. PloS Biol, 2004, 2(10):e354.
[14] Meyer CP, Paulay G. DNA barcoding:error rates based on comprehensive sampling[J]. PloS Biol, 2005, 3(12):e422.
[15] Elias M, Hill RI, Willmott KR, et al. Limited performance of DNA barcoding in a diverse community of tropical butterflies[J]. P Roy Soc B-Biol Sci, 2007, 274(1627):2881-2889.
[16] Lopez JV, Yuhki N, Masuda R, et al. Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat[J]. J Mol Evol, 1994, 39(2):174-190.
[17] Bensasson D, Zhang DX, Hewitt GM. Frequent assimilation of mitochondrial DNA by grasshopper nuclear genomes[J]. Mol Biol Evol, 2000, 17(3):406-415.
[18] Lu XM, Fu YX, Zhang YP. Evolution of mitochondrial cytochrome B pseudogene in genus Nycticebus[J]. Mol Biol Evol, 2002, 19(12):2337-2341.
[19] Kolokotronis SO, Macphee RD, Greenwood AD. Detection of mitochondrial insertions in the nucleus(NuMts)of Pleistocene and modern muskoxen[J]. BMC Evol Biol, 2007, 7:67.
[20] Pamilo P, Viljakainen L, Vihavainen A. Exceptionally high density of NUMTs in the honeybee genome[J]. Mol Biol Evol, 2007, 24(6):1340-1346.
[21] Black IV, WC, Bernhardt SA. Abundant nuclear copies of mitochondrial origin(NUMTs)in the Aedes aegypti genome[J]. Insect Mol Biol, 2009, 18(6):705-713.
[22] Thalmann O, Hebler J, Poinar HN, et al. Unreliable mtDNA data due to nuclear insertions:a cautionary tale from analysis of humans and other great apes[J]. Mol Ecol, 2004, 13(2):321-335.
[23] Song H, Buhay JE, Whiting MF, et al. Many species in one:DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified[J]. PNAS, 2008, 105(36):13486-13491.
[24] Moulton MJ, Song H, Whiting MF. Assessing the effects of primer specificity on eliminating numt coamplification in DNA barcoding:a case study from Orthoptera(Arthropoda:Insecta)[J]. Mol Ecol Resour, 2010, 10(4):615-627.
[25] Jin XB, Xia KL. An index catalogue of Chinese Tettigonioidea(Orthoptera:Grylloptera)[J]. Journal of Orthoptera Research, 1994, 3:15-41.
[26] 周志军, 杨明茹, 常岩林, 等.两种纺织娘线粒体基因组的比较分析[J]. 昆虫学报, 2013, 56(4):408-418.
[27] Folmer O, Black M, Hoeh W, et al. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates[J]. Mol Mar Biol Biotechnol, 1994, 3(5):294-299.
[28] Burland TG. DNASTAR’s Lasergene sequence analysis software[J]. Methods Mol Biol, 2000, 132:71-91.
[29] Thompson JD, Gibson TJ, Plewniak F, et al. The CLUSTAL_X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools[J]. Nucleic Acids Res, 1997, 25(24):4876-4882.
[30] Tamura K, Peterson D, Peterson N, et al. MEGA5:molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods[J]. Mol Biol Evol, 2011, 28(10):2731-2739.
[31] Cheng S, Higuchi R, Stoneking M. Complete mitochondrial genome amplification[J]. Nat Genet, 1994, 7(3):350-351.
[32] Collura RV, Auerbach MR, Stewart CB. A quick direct method that can differentiate expressed mitochondrial genes from their nuclear pseudogenes[J]. Curr Biol, 1996, 6:1337-1339.
[33] Sorenson MD, Quinn TW. Numts:a challenge for avian systematics and population biology[J]. The Auk, 1998, 115(1):214-221.
[34] 王继文.动物线粒体假基因的识别及其在进化生物学中的应用[J]. 动物学杂志, 2004, 39(3):103-108.
[35] Chung WK, Steiper ME. Mitochondrial COII introgression into the nuclear genome of Gorilla gorilla[J]. Int J Primatol, 2008, 29(5):1341-1353.
[36] Zhang DX, Godfrey MH. Nuclear integrations:challenges for mitochondrial DNA markers[J]. TREE, 1996, 11(6):247-251.
