线粒体脑肌病伴高乳酸血症和卒中样发作综合征一例报告
2014-01-11熊葶李尊波罗国刚刘建军王朝霞沈定国
熊葶,李尊波,罗国刚,刘建军,王朝霞,沈定国
·个案报道·
线粒体脑肌病伴高乳酸血症和卒中样发作综合征一例报告
熊葶1,2,李尊波2,罗国刚3,刘建军2,王朝霞4,沈定国2
线粒体脑肌病伴高乳酸血症和卒中发作综合征;影像学;肌肉病理学;线粒体DNA A3243G突变
线粒体脑肌病伴高乳酸血症和卒中样发作综合征(Mitochondrial myopathy,encephalopathy,lactic acidosis,and stroke-like episode,MELAS)为最常见的线粒体脑肌病,是线粒体DNA(mitochondrial DNA,mtDNA)及核DNA基因突变导致蛋白合成障碍引起的多器官、多组织的进行性变性疾病,临床表现为卒中样发作、脑病、肌病、乳酸性酸中毒及耳、眼、心、肾、胃肠道、内分泌等多系统损害[1-3]。本病累及器官广泛,神经系统症状多样,症状复杂,常易误诊、漏诊。本文报道1例如下。
1 临床资料
患者,女,25岁,农民,因“发作性四肢抽搐伴意识丧失4年,发作性视力下降1年半”于2010年1月20日就诊于我院。患者于4年前(2005年11月29日)因头昏、发热在当地就诊时突发头向后仰、双眼上翻、四肢强直、阵挛,颜面及口唇发绀,呼之不应,约2~3 min后抽搐停止,每10余分钟发作1次,发作间期意识不清,无舌咬伤,无大、小便失禁,给予地西泮(剂量不详)肌注后抽搐再未发作,当地医院诊断为“脑炎”,抗癫痫治疗2 d仍未清醒,期间因“呼吸不规律”给予呼吸机辅助呼吸,并于2005年12月1日第1次转入我院就诊。当时查体:体温38.0℃,呼吸30次/分,脉搏130次/分,血压 124/ 80mmHg(1 mmHg=0.133 kPa)。双肺呼吸音粗,可闻及湿性啰音。心率130次/分,律齐,各瓣膜听诊区未闻及病理性杂音。意识不清,双侧瞳孔等大等圆,对光反射灵敏,鼻唇沟对称,四肢肌张力低,疼痛刺激可见躲避动作,腱反射(+),双侧Babinski征、Chaddock征阴性,颈部似有抵抗,膝关节不能伸直。行腰穿后压力及脑脊液化验正常,肌酸激酶(creatine kinase,CK)290 U/L,头颅CT见右侧基底核区点状低密度灶。治疗2 d后意识清醒,查体:神志清,言语无困难,双瞳孔等大等圆,对光反射灵敏,眼球各方向活动自如,闭目有力,双侧鼻唇沟对称,声音嘶哑,咽反射存在,伸舌居中,舌肌无萎缩,肩关节活动受限,指关节过伸,拇指蹼小,膝关节活动受限,右足内翻,足趾关节屈曲畸形,脊柱后突、侧弯,髂腰肌力弱,痛觉正常,病理反射未引出。追问病史,母孕期正常,足月顺产,出生后无窒息史,出生后40 d曾发生1次高热惊厥。否认头部外伤史,否认一氧化碳中毒史,否认进食“米猪肉史”。其父母亲、一兄一弟均体健。幼时跑跳、走路跟不上同龄人,上学时不能参加正常的体育锻炼。拟诊“线粒体脑肌病”,因家属拒绝未能行肌肉活检。2年后(2007年12月12日)患者再次因癫痫发作来我院就诊,形式同前。复查CK 394 U/L,24 h动态脑电图可见广泛慢波,未见癫痫波发放;头颅MRI中见双侧基底核区陈旧性腔隙样病灶,见图1;简易智能精神状态检查量表(mini-mental state examination,MMSE)得分:21分。2年半后(2008年7月)突发双眼视力丧失,持续2 d后自诉“恢复正常”,未到医院就诊。4年后(2009年12月29日)出现头顶部闷痛,疼痛剧烈,伴呕吐,当地医院给予“鲁米那2支”肌注后入睡,3 h后醒来时发现双眼视力丧失,当地医院给予治疗(具体不详)后视力略好转,并诉“语言缓慢,反应慢,听力减退”,遂转入我院。查体:身高160 cm,神志清楚,反应迟钝,难以交流,检查不合作;双瞳孔等大等圆,对光反射灵敏,双眼仅能数指,双眼球居中,双侧鼻唇沟对称,听力减退,肩关节活动受限,指关节过伸,拇指蹼小,膝关节活动受限,右足内翻,足趾关节屈曲畸形,脊柱后突、侧弯,髂腰肌力弱,痛觉正常,四肢腱反射(++),病理反射未引出。
患者入院后,于清晨空腹时安静卧床0.5 h后抽静脉血查血乳酸。结果显示,静息空腹血乳酸6.21 mmol/L(正常范围0.5~2.2 mmol/L)。使用PHILIPS Achieva 1.5T磁共振机进行脑MRI平扫及头颅MRA检查。头颅MRI显示双侧颞、顶、枕叶长T1、长T2信号,T2FLAIR序列亦呈高信号,DWI序列双侧基底核区陈旧性病灶呈低信号,双侧颞叶、右侧顶、枕叶病灶沿皮质分布呈高信号,脑萎缩,见图1。头颅MRA见左侧颈内动脉虹吸部狭窄,余血管走行正常,未见血管闭塞。
患者知情同意后,在局部麻醉下行右侧肱二头肌活检术,肌肉标本分为2部分,一部分经液氮冷却及异戊烷速冻,制成10 μm冰冻切片,行常规组织学和酶组织化学染色,包括HE染色、改良Gomori染色(MGT)、还原性辅酶I染色(NADH-TR)、糖原染色、油红“O”染色、ATP酶(pH4.3、4.45、4.5、11.0、11.05、11.1、11.15)染色;另一部分在2.5%戊二醛中固定,行透射电镜观察。肌肉组织化学染色结果显示,HE染色见肌纤维大小基本一致,无炎细胞浸润;MGT染色可见破碎红纤维;选取3个不同视野,每个视野计数100个纤维,计数破碎红纤维数量,结果显示破碎红纤维数量可达3%,见图2;HE、NADH-TR亦可见破碎红纤维;糖原染色未见糖原增多;油红“O”染色示少数肌纤维内脂滴稍多;ATP酶示Ⅰ、Ⅱ型纤维镶嵌分布,未见群组化现象。透射电镜下观察超微结构显示,肌原纤维破坏,Z线结构不清,呈水波纹样改变,有脂滴堆积,肌膜下及肌原纤维间大量线粒体堆积,线粒体大小、性状不一,并可见类晶格状包涵体,见图2。

图1 患者头颅MRI图像
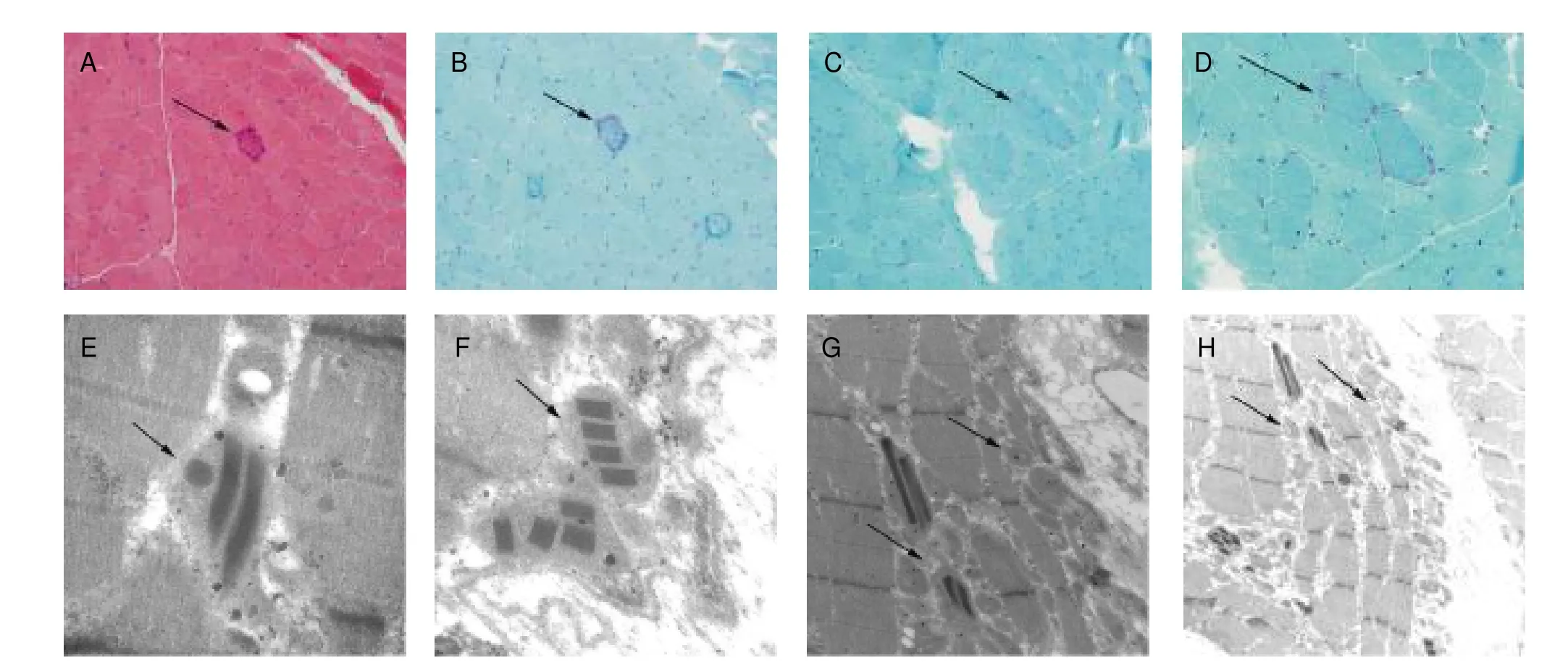
图2 患者肌肉病理
北京大学第一医院神经内科、神经病理研究室进行mtDNA检查显示,mtDNA A3243G点突变,见图3。

图3 患者mtDNA A3243G位点存在致病突变
2 讨论
本例为25岁女性,主要临床特点为反复癫痫发作、发作性视力减退、伴听力障碍,自幼有运动不耐受,伴关节畸形(手指指间关节过伸),慢性智能减退;静息空腹血乳酸增高,头颅 MRI-T2、DWI、T2-FLAIR可见双侧枕叶、顶叶、颞叶有片状病变,伴脑萎缩;肌肉病理示有明确的破碎红纤维;电镜可见线粒体累积及类晶格样包涵体;线粒体DNA测定有mtDNA3243突变,可明确诊断为MELAS。
MELAS的核心症状是卒中样发作(stroke-like episodes,SLEs),表现为急性突发性或复发性脑神经功能障碍,具有相应的影像学改变[1-5],由于损害部位不同可有不同临床症状及其症状组合,可表现为视力障碍、偏瘫、语言障碍、精神及其它局灶性神经症状。本例出现发作性头痛、呕吐,发作性视力障碍,影像学可见双侧枕叶、颞叶、顶叶的水肿样改变。本例有2次以癫痫发作为特点,表现为全面性强直-阵挛发作及癫痫持续状态。癫痫可以出现在卒中样发作阶段,本例1次癫痫发作后头颅CT表现为双侧基底核区点状低密度灶,另一次癫痫发作后头颅MRI见到基底核区陈旧性腔隙样病灶,但DWI及T2未见到典型水肿改变,提示癫痫也可出现在MELAS的不同病程中。
本例影像学改变还发现双侧颞、顶、枕叶几乎对称的长T1长T2片状改变,DWI主要累及右侧皮质,呈脑回样分布,有作者称为“层状坏死”,双侧的不同病变推测为病变时期不同所致,影像学改变与临床症状的视、听障碍关联。文献中报告好发部位为脑的后部,枕叶、顶叶或颞叶,少数可累及额叶,主要累及皮质,少数也可累及白质,表现为水肿样病灶,T2加权成像、DWI及FLAIR像呈高信号[6]。SLLs(stroke-like lesion)的局部水肿可在短时间内消退,少数病例可伴出血。本例的末次影象学改变符合上述特征,但因未复查未能见到病灶消退。MELAS颅脑MRI影像尚有小的散发病灶出现在基底核区,可见基底核坏死或钙化。本例的CT及1次因癫痫发病的MRI改变说明同一患者不同时期、不同发作形式影像可有差异。
目前认为MELAS的SLEs并非血管源性,而是代谢引起的脑水肿,SLLs病灶分布范围常超过血管供血范围。本例头颅MRA虽可见左侧颈内动脉虹吸部狭窄,未见明显血管闭塞,其余血管走行正常,双侧脑叶累及,病变范围超过单一血管供血范围,也佐证了上述观点。脑萎缩多见于亚急性期、慢性期,本例患者除脑萎缩外,MMSE 21分,智能下降,精神行为有一定异常,可能与反复发作或神经损害的进行性累积造成最终的神经功能持久性障碍有关[7]。
Hirano等[2]综合文献分析110例MELAS患者中肌力弱出现率可达89%,但应强调部分患者缺乏肌肉力弱、肌肉病理缺乏典型改变,增加了疾病诊断难度,需要进行综合分析,才能做出正确诊断。本例尚有听力减退,也为MELAS的常见症状,文献报告发生率达77%[8]。另外,本例表现为多处关节畸形,如脊柱后突、侧弯,双手指间关节过伸、膝关节挛缩、双肩关节活动受限,拇指蹼小,右足内翻,复习文献中尚未见到相关报告。肌肉病理除见到线粒体异常外,未见到肌纤维结构异常或肌纤维分型异常,不支持同时存在有先天性肌肉疾病,其真正原因尚待进一步观察。本例经mtDNA测定证实有mtDNA A3243G突变。mtDNA突变检测是诊断本病的最重要技术[2-3],MELAS患者的基因具有明显的基因异质性,目前已发现多种基因突变。80%MELAS患者常见mtDNA A3243G突变,其次为T3271C突变,占7.5%,此外还有3291T>C、10191 T>C、G1246A、T8316C突变等[3]。但并非所有MELAS患者都可检出基因突变[9],而mtDNA A3243G点突变也可表现为非MELAS的线粒体病,如母系遗传性糖尿病和耳聋、线粒体心肌病以及无症状的基因携带者等,因此具有临床异质性[10]。
目前对MELAS仍缺乏特效治疗,一些药物的治疗目的为改善线粒体电子传递链的功能,扩张小血管,减轻血管性水肿,可选用L-精氨酸、辅酶Q10、艾地苯醌、肉碱等线粒体基质及辅助因子。也可联合应用其他药物,如维生素E、依达拉奉、细胞色素C。本病预后较差,必须进一步探索新的有效治疗。
[1]沈定国.线粒体脑肌病伴高乳酸血症和卒中样发作综合征的临床诊断治疗再认识[J].中华神经科杂志,2011,44:304-308.
[2]Hirano M,Pavlakis SG.Mitochondrial myopathy,encephalopathy,lactic acidosis,and strokelike episodes(MELAS):current concepts [J].J Child Neurol,1994,9:4-13.
[3]Finsterer J.Genetic,pathogenetic,and phenotypic implications of the mitochondrial A3243G tRNA mutation[J].Acta Neurol Scand, 2007,116:1-14
[4]Testai FD,Gorelick,PB.Inherited metabolic disorders and stroke Part 1,Fabry disease and mitochondrial myopathy,encephalopathy,lactic acidosis and strokelike episodes[J].Arch Neurol, 2010,67:19-24.
[5]Finsterer J,Stollberger C.Stroke in myopathies[J].Cerebrovascular Dis,2010,29:6-13.
[6]Finsterer J.Central nervous system imaging in mitochondrial disorders[J].Can J Neurol Sci, 2009,36:143-153.
[7]Salsano E,Giovagonoli AR,Morandi L.Mitochondrial dementia:a sporadic case of progressive cognitive and behavioral decline with hearing loss due to the rare m.3291T>C MELAS mutation[J].Neurol Sci,2011,300:165-168.
[8]Kaufman KR,Engelstad K,Wei Y,et al.Protean phenotypic features of the A3243G mitochondrial DNA mutation[J].Arch Neurol,2009, 66:85-91.
[9]Abu-Amero KK,Al-Dhalaan H,Bohlega S.A patient with typical clinical features of mitochondrial encephalopathy, lactic acidosis and stroke-like episodes(MELAS)but without an obvious genetic cause:a case report[J].Med Case Report,2009,3:77-81.
[10]Maassen JA,van den Ouweland JM,t Hart LM,et al.Maternally Inherited Diabetes and Deafness:A diabetic subtype associated with a mutation in mitochondrial DNA[J].Horm Metab Res,1997,29:50-55.
(本文编辑:唐颖馨)
R741;R746.9
A DOI 10.3870/sjsscj.2014.04.028
1.西安交通大学医学院西安710061 2.西安高新医院神经内科西安710075 3.西安交通大学医学院第一附属医院神经内科西安710061 4.北京大学第一医院神经内科北京100034
2014-03-17
刘建军ljjfzc57@vip.sina.com
