三有机锡(4R)-3-[[(2S)-5-氧-2-吡咯烷基]羰基]-4-噻唑烷甲酸酯的合成、结构和体外抗癌活性
2014-01-02田来进王晓龙郑晓亮董云赛刘志坤
田来进 王晓龙 郑晓亮 董云赛 刘志坤
(1曲阜师范大学医药中间体与天然药物山东省高校重点实验室,曲阜 273165)
(2浙江省医学科学院药物研究所,杭州 310013)
有机锡化合物作为一类重要的有机金属化合物被广泛用作反应催化剂、PVC热稳定剂、杀菌防腐剂和防污涂料[1]。近年发现许多有机锡化合物具有良好的抗癌活性,而且其活性与锡原子上的烃基类型、锡原子配位数及配体分子密切相关[2-5]。因此,将具有特定结构和生物功能的配体与有机锡基团结合起来是获取高活性有机锡化合物的重要途径。通过这一途径合成的环戊烷甲酸、氨基酸、吡啶二甲酸、联苯乙酸、扑酸、呋喃甲酸等羧酸的有机锡酯都具有多样的结构和良好的体外抗癌活性[4-9]。(4R)-3-[[(2S)-5-氧-2-吡咯烷基]羰基]-4-噻唑烷甲酸(匹多莫德)是一种人工合成的二肽,临床上主要用于预防和治疗反复性呼吸道感染、慢性支气管炎、过敏性鼻炎和哮喘,还可用于多种病毒感染和恶性肿瘤等造成的机体免疫功能低下[10-11]。目前为止,尚未见文献报道匹多莫德金属配合物的结构和生物活性的研究。为继续扩展有机锡化合物的结构类型及抗癌活性的筛选,本文将匹多莫德引入三烃基锡分子中合成了具有右螺旋结构的三烃基锡(4R)-3-[[(2S)-5-氧-2-吡咯烷基]羰基]-4-噻唑烷甲酸酯(1),并测定了其体外抗癌活性。
1 实验部分
1.1 试剂和仪器
三环己基氢氧化锡和三苯基氢氧化锡购自Aldrich公司,三(2-甲基-2-苯基丙基)氢氧化锡和(4R)-3-[[(2S)-5-氧-2-吡咯烷基]羰基]-4-噻唑烷甲酸(HL)分别按文献[12-13]方法制备,L-半胱氨酸为生化试剂,其它试剂和溶剂为市售分析纯试剂。所用仪器为WRS-1A数字熔点仪,PE 2400 Series II元素分析仪,Nicolet NEXUS-470 FT-IR谱仪 (KBr压片),Bruker Avance 300核磁共振仪 (TMS作内标,CDCl3为溶剂)。
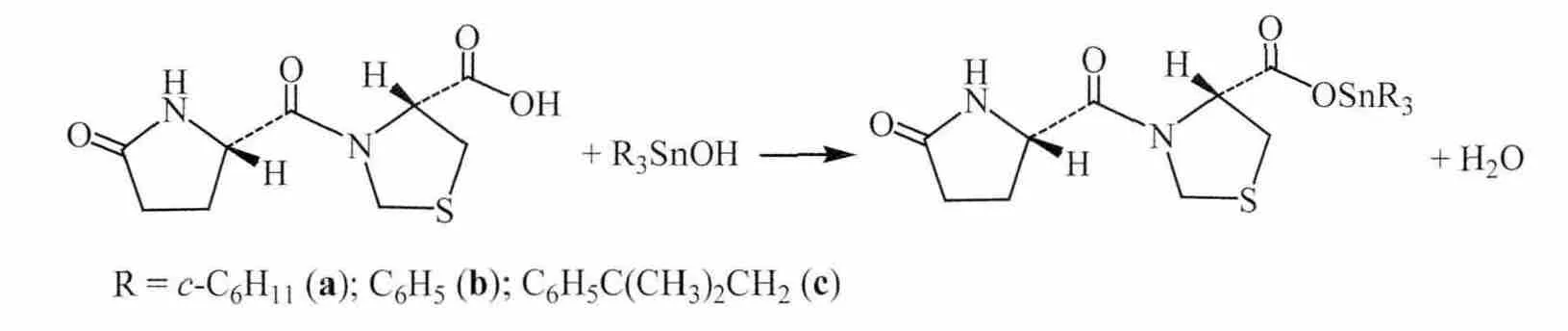
1.2 三有机锡(4R)-3-[[(2S)-5-氧-2-吡咯烷基]羰基]-4-噻唑烷甲酸酯的合成
在100 mL圆底烧瓶中,分别加入配体HL(0.49 g,2 mmol)、三环己基氢氧化锡(0.77 g,2 mmol)或三苯基氢氧化锡(0.73 g,2 mmol)或三(2-甲基-2-苯基丙基)氢氧化锡(1.07 g,2 mmol)、45 mL 苯和 15 mL 乙醇,装上Dean-Stark分水器,加热回流反应6 h,趁热过滤,滤液用旋转蒸发仪除去溶剂,得淡黄色固体,用乙醇重结晶,得无色晶体。反应式如下:
三环己基锡(4R)-3-[[(2S)-5-氧-2-吡咯烷基]羰基]-4-噻唑烷甲酸酯(1a):收率 83%,m.p.153~154 ℃。元素分析按 C27H44N2O4SSn,实测值(%):C,53.06;H,7.13;N,4.55。计算值(%):C,53.04;H,7.25;N,4.58。IR(KBr,cm-1):3 234(N-H),1 695[ν(COO-)as)],1 652,1 583(C=O),1 417[ν(COO-)s]。1H NMR(CDCl3)δ:6.01(s,1H,NH),4.93~4.27(m,4H),3.55~3.25(m,2H,SCH2),2.49~2.15(m,4H,COCH2CH2),1.95~1.29(33H,m,c-C6H11)。
三苯基锡(4R)-3-[[(2S)-5-氧-2-吡咯烷基]羰基]-4-噻唑烷甲酸酯(1b):收率 78%,m.p.172~173 ℃。元素分析按 C27H26N2O4SSn,实测值 (%):C,54.29;H,4.13;N,4.59。计算值(%):C,54.66;H,4.42;N,4.72。IR(KBr,cm-1):3 230(N-H),1 690[ν(COO-)as)],1 650,1 579(C=O),1 415[ν(COO-)s]。1H NMR(CDCl3)δ:7.68(6H,dd,J=2.1,7.5 Hz,3J(119Sn-H)=62.2 Hz,o-H of C6H5),7.41~7.34(9H,m,m-H and p-H of C6H5),6.09(s,1H,NH),4.85~4.23(m,4H),3.44~3.19(m,2H,SCH2),2.43~2.07(m,4H,COCH2CH2)。
三(2-甲基-2-苯基丙基)锡(4R)-3-[[(2S)-5-氧-2-吡咯烷基]羰基]-4-噻唑烷甲酸酯(1c):收率68%,m.p.59~60 ℃。 元素分析按 C39H50N2O4SSn,实测值(%):C,61.66;H,6.49;N,3.58。计算值(%):C,61.50;H,6.62;N,3.68。 IR(KBr,cm-1):3 239(N-H),1 697[ν(COO-)as)],1 655,1 608(C=O),1 406[ν(COO-)s]。1H NMR(CDCl3)δ:7.31~7.20(9H,m,m-H and p-H of C6H5),7.16(6H,d,J=7.4 Hz,o-H of C6H5),6.10(1H,s,NH),4.90~4.25(m,4H),3.55~3.25(m,2H,SCH2),2.48~2.14(m,4H,COCH2CH2),1.29(18H,s,6CH3),1.25(6H,s,J(119Sn-H)=52 Hz,CH2Sn)。
1.3 晶体结构测定
将HL、1a和1b的乙醇溶液于室温下慢慢挥发,得到无色单晶。选取尺寸适宜的晶体置于Bruker Smart Apex单晶衍射仪上,以石墨单色化的Mo Kα 辐射(λ=0.071 073 nm)为光源,以 φ-ω 扫描方式,在295(2)K收集衍射数据。晶体结构由直接法解出,非氢原子坐标是在以后的数轮差值Fourier合成中陆续确定的。基于F2以全矩阵最小二乘法对全部非氢原子坐标及各向异性热参数进行精修,氢原子坐标由理论计算加入。结构解析及修正的全部计算用SHELX-97程序完成。化合物的晶体数据和结构精修列于表1。
CCDC:837609,HL;837610,1a;837611,1b。
1.4 体外抗癌活性测定
将1a和1b溶于DMSO,准确配制200 mg·L-1的存储液,临用前以无血清DMEM培养液稀释至所需浓度(DMSO终浓度小于0.1%)。人结肠癌Colo205细胞及人乳腺癌Bcap37细胞购自中科院上海细胞库,用含10%新生牛血清、40 U·mL-1庆大霉素的DMEM培养液,于37℃、5%CO2和饱和湿度的培养箱中传代培养,取对数生长期细胞用于实验。化合物的体外抗癌活性用MTT法测定[14],不同浓度受试药物对肿瘤细胞的增殖抑制率经计算处理得到半数抑制浓度IC50[15]。
2 结果与讨论
2.1 波谱分析
在配合物1a~1c的红外光谱中,配体分子在2 650~3 400 cm-1的 ν(OH)特征吸收峰消失,表明羧基质子被烃基锡取代而形成了配合物,这被出现在525 cm-1附近的Sn-O键的伸缩振动进一步证实。3 230~3 239 cm-1处的尖峰归于NH键的伸缩振动,1 650~1 655 cm-1和 1 579~1 608 cm-1的吸收峰归于配合物分子中的酰胺和内酰胺羰基的伸缩振动。和配体酰胺羰基(1 656 cm-1)相比变化很小,而内酰胺的羰基(1 623 cm-1)则向低波数分别移动了40、44和15 cm-1,表明在1a~1c中均存在内酰胺羰基氧对Sn 原子的配位作用[16],其作用强度按 1b、1a、1c 依次减弱,这和锡原子上的烃基体积依次增大是一致的。配合物1a~1c的羧基不对称伸缩振动ν(COO)as和对称伸缩振动ν(COO)s分别出现在1 690~1 697和1 406~1 417 cm-1,二者之差分别为278、275和291 cm-1,表明羧基是以单齿形式和锡原子结合[17]。

表1 HL、1a和1b的晶体数据和结构精修Table 1 Crystallographic data and structure refinements of HL,1a and 1b
在配合物的1H NMR谱中,没有观察到配体酸中羧基的质子峰,进一步证明羧基质子被烃基锡取代。配合物分子中杂环上2个手性碳原子的存在使得环上质子出现较为复杂的共振吸收峰,内酰胺氮质子在6.10处出现单峰,吡咯烷酮环上COCH2CH2质子在2.10~2.50出现多重峰,噻唑烷环上的SCH2质子在3.25~3.55范围出现多重峰,杂环上其他4个质子在3.25~3.55范围出现多重峰。与锡相连的烃基质子均出现在正常的位置[7-9]。
2.2 化合物的晶体结构
手性配体HL结晶在正交晶系的P212121手性空间群,其分子结构见图1。分子的羧基C-O键(C(1)-O(1)0.119 0(2),C(1)-O(2)0.129 7(2)nm)和酰胺 C-N键(C(5)-N(1)0.134 9(2),C(9)-N(2)0.135 4(2)nm)键长均在正常范围,2个五元杂环呈信封式构象。分子间通过羧基与内酰胺形成的氢键O(2)-H(2)…O(4)#1(对称操作 #1:x-1/2,-y-1/2,-z)和 N(2)-H(2A)…O(1)#2(对称操作 #2:-x+5/2,-y,z-1/2)连接成三维超分子结构。

图1 配体HL的分子结构(30%概率水平)Fig.1 Molecular structure of HL with ellipsoids at the 30%probability level

图2 1a中锡原子的配位环境(30%概率水平)Fig.2 Coordination environment of the tin atom in 1a with ellipsoids at the 30%probability level

表2 配合物1a和 1b的重要键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and angles(°)of 1a and 1b
1a和1b的空间群分别为P212121和P21手性空间群,中心Sn原子的配位环境如图2和图3所示,重要键长和键角列于表2。1a的中心锡原子是5配位的,5个配位原子分别来自3个环己基碳(C(1)、C(7)和C(13))、配体的羧基氧(O(1))以及邻近另一分子配体的内酰胺羰基氧(O(4A))(对称操作A:x+1/2,-y+3/2,-z),围绕锡原子排列为畸变的三角双锥构型,3个碳原子占据了三角双锥赤道平面的3个位置,3个Sn-C键长几乎相等,绕锡原子的3个键角分别为 115.71(10)°、117.57(10)°和 122.72(9)°,三者之和为356.00(29)°,锡原子向羧基O(1)方向偏离赤道平面0.025 1(3)nm;2个氧原子O(1)和O(4A)占据了三角双锥的轴向位置,键角O(1)-Sn(1)-O(4A)为174.05(7)°,2个 Sn-O键的键长分别为 Sn(1)-O(1)0.215 88(16)和 Sn(1)-O(4A)0.267 5(2)nm,显然羧基氧O(1)与锡的键合比内酰胺羰基氧O(4A)对锡的配位强的多。在晶体中,配体分子的这种桥联配位将1a连接为一维右螺旋链状聚合物 (见图4),相邻2个锡原子间的距离为0.796 1(2)nm,相间2个锡原子间的距离为1.030 2(2)nm。羧基的C(19)-O(1)和C(19)-O(2)键长分别为0.128 9(3)nm和0.121 8(3)nm,具有明显的单、双键特征,进一步证实了羧基的单齿配位模式。

图3 1b中锡原子的配位环境(30%概率水平)Fig.3 Coordination environment of the tin atom in 1b with ellipsoids at the 30%probability level
1b的结构和1a相似,也是通过手性配体的羧基氧O(1)和内酰胺羰基氧O(4A)(对称操作A:-x+2,y-1/2,-z+1)对锡桥联配位形成的五配位一维右螺旋链状结构(见图4)。1b的三角双锥赤道平面内的3个Sn-C键的键长(0.213 0(3)~0.211 5(2)nm)和报道的五配位三苯基锡化合物Ph3Sn(O2CCH2NHCONH2)相 似[16],3 个 C-Sn-C 键 角 分 别 为 124.37(16)°、110.72(11)°和 122.46(14)°,三者之和为 357.55(41)°,最大与最小键角之差(13.65(5)°)明显大于 1a(7.01(1)°)。锡原子向羧基O(1)方向偏离赤道平面0.019 2(3)nm,2个三角双锥轴向位置的Sn-O键键长分别为Sn(1)-O(1)0.212 8(2)和 Sn(1)-O(4A)0.246 2(2)nm,后者明显比1a短(差值0.021 3 nm),表明在1b中内酰胺羰基的配位作用比1a强,这也反映在-C(=O)NH-中C=O和C-N键键长的变化上,1b的C=O键长比1a的长,而C-N键长则比1a的短(见表2)。这一结果与1a和1b内酰胺羰基的红外吸收变化是一致的。在右螺旋链中,相邻2个锡原子间的距离为0.872 3(2)nm,相间2个锡原子间的距离为1.065 1(2)nm,均比1a的距离长。

图4 1a和1b的一维右螺旋链Fig.4 1D right-hand helical chain of 1a and 1b
2.3 体外抗癌活性
化合物1a和1b对人结肠癌CoLo205细胞和人乳腺癌Bcap37细胞增殖均有强的抑制作用,其半数抑制浓度 IC50分别为 0.82±0.10、0.52±0.03(CoLo205) 和 0.32±0.07、0.16±0.01(Bcap37)μmol·L-1,活性均高于临床上使用的金属配合物抗癌药物顺铂 (IC5013.74±0.39 和 5.93±0.83 μmol·L-1),1a 对这2种癌细胞的活性均高于1b。这一结果与文献报道的三环己基和三苯基锡4-联苯-4-丁酮酸酯[18]、三环己基锡呋喃甲酸酯[9]相近,为进一步研究其生物活性提供了重要参考。
[1]Davies A G,Gielen M,Pannell K H,et al.Tin Chemistry:Fundamentals,Frontiers,and Applications.Weinheim:John Wiley&Sons,2008:413-719
[2]Gielen M,Biesemans M,Willem R.Appl.Organometal.Chem.,2005,19:440-450
[3]Hadjikakou S K,Hadjiliadis N.Coord.Chem.Rev.,2009,253:235-249
[4]Shang X,Meng X,Alegria E C B,et al.Inorg.Chem.,2011,50:8158-8167
[5]TIAN Lai-Jin(田来进),YANG Hong-Jun(杨红军),ZHENG Xiao-Liang(郑 晓 亮),et al.Chinese J.Inorg.Chem.(无 机 化学学报),2009,25:629-632
[6]Garcia-Zarracino R,Hopfl H.J.Am.Chem.Soc.,2005,127:3120-3130
[7]TIAN Lai-Jin(田来进),SUN Yu-Xi(孙玉希),ZHENG Xiao-Liang(郑晓亮),et al.Chinese J.Inorg.Chem.(无机化学学报),2006,22:2031-2036
[8]ZHANG Jun-Hong(张军红),ZHANG Ru-Fen(张茹芬),MA Chun-Lin(马春林),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30:664-670
[9]YU Jiang-Xi(庾江喜),FENG Yong-Lan(冯泳兰),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30:1267-1272
[10]Riboldi P,Gerosa M,Meroni P L.Int.J.Immunopat.Ph.,2009,22:255-262
[11]TIAN Xin-Ping(田新平),ZENG Xiao-Feng(曾小峰).Chin.J.New Drugs(中国新药杂志),2005,14:111-114
[12]Reichle W T.Inorg.Chem.,1966,5:87-91
[13]HU Kun(胡昆),WANG Zhen-Shan(王振山),REN Jie(任杰).Chin.J.Pharm.(中国医药工业杂志),2009,40:883-884
[14]Denizot F,Lang R.J.Immunol.Methods,1986,89:271-275
[15]Zheng X L,Sun H X,Liu X L,et al.Acta Pharmacol.Sin.,2004,25:1090-1095
[16]Kamruddin S K,Chattopadhyaya T K,Roy A,et al.Appl.Organometal.Chem.,1996,10:513-521
[17]Deacon G B,Phillips R J.Coord.Chem.Rev.,1980,33:227-250
[18]Tian L,Yu Q,Zheng X,et al.Appl.Organometal.Chem.,2005,19:672-676
