金属离子导向自组装的磺酰化对氨基苯甲酸配合物的合成与表征
2014-01-02黄妙龄翁文婷
黄妙龄 翁文婷 陈 强
(泉州师范学院化学与生命科学学院,化工轻工清洁生产技术协同中心,泉州 362000)
0 引 言
金属离子导向的分子自组装研究是配位超分子化学的一个重要的组成部分。根据金属离子配位构型的需求,有机配体的结构和反应条件等诸多因素,人们已经合成了一系列如螺旋、分子梯、墙砖式等一维,二维,三维网状配位聚合物[1-10]。研究表明:分子自组装的形式和金属配体的构型主要由中心原子的配位构型和有机配体的结构所决定。当然,金属离子和配体的摩尔比及反应条件也可影响配位聚合物的构型。另外,金属离子本身不仅可以作为配位聚合物网络结构中的节点,还可以通过某些桥联配体或者桥连原子的作用形成特定的SBUs(次级构筑单元)。而这些SBUs可以进一步作为节点来构筑网络结构[11-13]。因此,选择合适的具有特定官能团的有机合成子和桥联金属离子构建特定的功能材料已在许多交叉学科中得到快速发展,如晶体工程、配位化学、超分子化学和材料科学等[14-16]。本文通过芳香羧酸的磺酰化衍生物与含氮桥螯合配体菲咯啉和相应的过渡金属盐在常规溶液中自组装反应,得到了4个配位化合物,并对它们的电化学和荧光性质进行了研究。
1 实验部分
1.1 试剂与仪器
N-对甲苯磺酰对氨基苯甲酸为自制药品,其它所有试剂均为分析纯,使用时未进行纯化。美国Nicolet iS10型FT-IR红外光谱仪(KBr压片),光谱范围 4 000~400 cm-1;德国 NETZSCH STA 409 PC型综合热分析仪 (在N2气氛中,升温速率10℃·min-1);德国Bruker SMART APEXⅡCCD单晶衍射仪;国产CHI660d电化学工作站;美国VARIAN Cary/Eclipse荧光分光光度仪。
1.2 配体N-对甲苯磺酰对氨基苯甲酸的合成
对甲苯磺酰氯与对氨基苯甲酸按1:1的物质的量比混合,置于500 mL的烧杯中,加入适量的蒸馏水,用NaOH调节至pH=10,同时在冰水浴中反应1 d,过滤。滤液用浓HCl调至pH=2,产生白色沉淀,抽滤,得白色固体,放入烘箱干燥20 h后,研磨至粉末备用。
1.3 配合物1~4的合成
配合物1到4的合成方法均相似,只是过渡金属盐不同。取1mmol的N-对甲苯磺酰对氨基苯甲酸溶于7 mL的N,N-二甲基甲酰胺 (DMF),称取1mmol的菲咯啉溶于5mL的乙醇,再称取1 mmol的过渡金属盐溶于5 mL的水中(其中金属盐:配合物1为氯化铜、配合物2为氯化锌、配合物3为醋酸铅、4为硝酸铅),将以上3种溶液混合于圆底烧瓶,用氢氧化钠调节pH=5左右,于70℃左右反应12 h。冷却至室温,过滤。滤液放置自然挥发。14 d后得蓝色块状晶体1,产率67%;15 d后得无色块状晶体2,产率53%;30 d后得无色块状晶体3和4,产率分别为 61%和 57%。配合物 1,IR(KBr,cm-1):3 528s、1 662s、1 607vs、1 551s、1 519m、1 393vs、1 322s、1 157vs、1 089vs、931m、848s、797s、723s、657 s、575m、546m、464w、418vw。 配 合 物 2,IR(KBr,cm-1):3 527s、1 650w、1 606vs、1 552s、1 518m、1 393 vs、1 333s、1 159vs、1 090vs、921s、848m、782s、726s、670s、577m、547m、469w、425vw。配合物 3,IR(KBr,cm-1):1 655vs、1 594s、1 530s、1 515w、1 383vs、1 336 s、1 160vs、1 091s、921s、847s、787s、722s、657s、580 m、548m、460w、417w。 配合物 4,IR(KBr,cm-1):1 607 s、1 591s、1 537vs、1 515w、1 385vs、1 334s、1 319m、1 152vs、1 087s、922s、850m、783s、727m、660s、576s、547m、462w、417vw。
1.4 配合物1~4晶体的X射线衍射
选取配合物1~4合适大小单晶体,置于Bruker SMART APEXⅡCCD衍射仪上,用石墨单色化的Mo靶 Kα 射线(λ=0.071 073 nm)以 φ-ω 扫描方式分别收集衍射数据。数据经经验吸收校正,晶体结构用SHELXS-97程序通过直接法解析,用SHELXL-97程序基于F2对全部非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法精修。配合物1~4的晶体学数据和结构精修参数列于列于表1,主要键长和键角列于表2,有关氢键的键长和键角列于表3。
CCDC:968877,1;824654,2;939749,3;939748,4。
2 结果与讨论
2.1 配合物的晶体结构
2.1.1 配合物[Cu(Ts-p-aba)2(phen)(H2O)]n(1)的晶体结构
配合物1为1D链状结构(图1a),其不对称单元包含1个Cu2+离子,2个Ts-p-aba配体,1个phen分子和1个配位水分子。有6个不同原子与中心Cu2+离子配位,其中 2 个 N 原子[N(3)、N(4)]来自phen分子,4个氧原子有3个来自Ts-p-aba配体,另1个则由配位水分子提供。Cu-N键长范围是 0.201 4(2)~0.203 3(3)nm;Cu-O 距离范围为0.194 5(2)~0.267 5(5)nm, 其 中 Cu-O(7)距 离 为0.238 2(2)nm,Cu-O(6)#1距离为 0.267 5(5)nm,相对Cu-O(3)和Cu-O(9)的距离拉长了许多,因此Cu2+离子的配位环境为拉长的八面体几何构型。O(7)和 O(6)#1占据轴向位置,∠O(6)#1-Cu(1)-O(7)=172.81(58)°;N(3)、N(4)、O(3)和 O(9)原子位于赤道位置,扭转角∠N(4)-O(3)-O(9)-N(3)=2.52°,接近共平面。配体Ts-p-aba在配合物中有2种配位模式:一种是以单齿羧基氧原子与Cu2+离子配位;另一种除了单齿羧基氧原子与Cu2+离子配位外,-SO2基上的氧原子也参与了配位,这2个氧原子与Cu2+离子的配位作用较弱,键长较长,使整个分子形成一维链。Cu…Cu距离为1.370 72(12)nm,Cu-Cu-Cu之间的夹角是180.000(4)°,说明一维链上的每个Cu2+离子都在同一直线上,phen分子的H原子与相邻链上主配体Ts-p-aba中未参与配位的O原子形成了CH…O 弱氢键(表 3),其中 C(36)-H(36)…O(4)将配合物 1 扩展为 2D 网格状(图 1b),C(30)-H(30)…O(1)进一步将配合物扩展为3D超分子结构(图1c)。

表1 配合物1~4的晶体学数据和结构精修参数Table 1 Crystal data and structure parameters of complexes 1~4

表2 配合物1~4的键长和键角Table 2 Selected Bond lengths(nm)and bond angles(°)for complexes 1~4

续表1

表3 配合物1~4的氢键键长和键角Table 3 Parameters of hydrogen bonds for the complexes 1~4

图1 配合物1的结构Fig.1 Structures of complex 1
2.1.2 配合物[Zn(Ts-p-aba)2(phen)(H2O)]·H2O(2)的晶体结构
配合物2是一个单分子配合物(图2a),由1个锌离子(Zn2+)、2 个 Ts-p-aba、1 个 phen、1 个配位水分子和1个结晶水组成的 (其中这个结晶水分子是由3个在晶胞中的占有率分别为0.3、0.3、0.4的水分子组成)。锌离子周围有5个原子与之配位。2个氮原子[N(1)、N(2)]来自于配体菲咯啉(phen),3 个氧原子[O(1W)、O(2)、O(4)]由配位水和配体 Ts-p-aba 的羧基氧原子提供。这5个原子在锌离子周围形成一个畸变的四角锥体的配位环境,其中O(2)在顶点,N(1)、N(2)、O(4)、O(1W)在底部,4 个原子接近共平面[扭转角为∠O(1W)-O(4)-N(2)-N(1)=5.56°]。 2 个 Ts-paba配体的羧基均以单齿方式与中心Zn2+离子配位,整个分子呈“人”字型。相邻配合物分子间通过phen分子的H原子与配体Ts-p-aba中未参与配位的氧原子及磺酰基(-SO2)上的氧原子形成了C-H…O弱氢键,将配合物2扩展为2D网格状(图2b)。

图2 配合物2的结构Fig.2 Structures of complex 2
2.1.3 配合物[Pb(Ts-p-aba)2(phen)]2(3)的晶体结构
配合物3是一个二聚体分子(图3a),由2个铅离子、2个phen以及4个Ts-p-aba组成。在配合物3中,铅离子为六配位,其中2个氮原子[N(2)、N(3)]来自于配体 phen;4 个氧原子[O(1)、O(2)、O(5)和 O(6)]由配体Ts-p-aba的羧基的氧原子提供。Ts-p-aba的2个氧原子(O1、O2)为双齿桥联模式,把相邻2个铅离子连接在一起,而另外1个配体上的2个氧原子[O(5)、O(6)]为双齿螯合模式。Pb(II)的配位多面体为畸变的八面体构型,O(2)-Pb(1)-N(4)键角为144.65°,位于八面体的轴向位置;O(1)、N(3)、O(5)、O(6)位于八面体的赤道平面,O(1)#1-O(5)-O(6)-N(3)的扭转角为6.08°,接近于共平面。配合物中还存在着分子间氢键 N(1)-H(1)…O(7)和 N(2)-H(2)…O(1),起着稳定分子的作用,且把配合物分子扩展为3D超分子网状结构(图 3b)。
2.1.4 配合物{[Pb(Ts-p-aba)(phen)(NO3)](DMF)}n(4)的晶体结构

图3 配合物3的结构Fig.3 Structures of complex 3
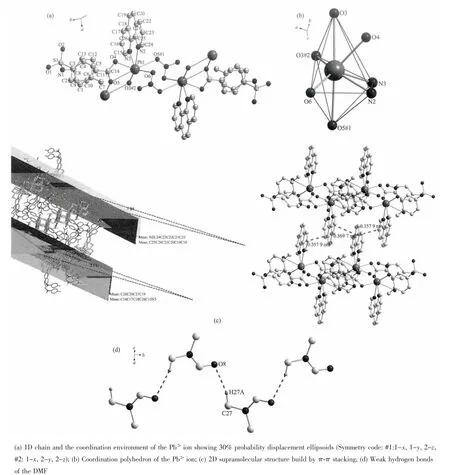
图4 配合物4的结构Fig.4 Structures of complex 4
配合物4是1D聚合链结构(图4a)。与配合物3相比,配合物4的结构要复杂些。其不对称单元包含1个铅离子 (Pb2+)、1个Ts-p-aba配体、1个phen分子、1个硝酸根和1个游离的DMF分子。中心Pb2+离子处于变形的一面心八面体配位环境中 (图4b),其中 O(3)和 O(5A)位于两极,O(3B)、O(6A)、N(2)和N(3)位于赤道上,扭转角∠O(3B)-O(6A)-N(2)-N(3)=-11.297(2)°,O(4)在 O(3)、N(2)和 N(3)组成的面上方。相邻两个Pb2+离子通过二齿桥联的硝酸根组成[Pb2O4N2]重复结构单元,每个[Pb2O4N2]结构单元再通过配体Ts-p-aba羧基上的O原子相互连接成1D岛形链结构。配体Ts-p-aba羧基氧原子的配位方式为螯合加单齿桥联。[Pb2O4N2]结构单元为中心对称,2个菲咯啉分别处于1D链的上下两侧。相邻分子间的菲咯啉的吡啶环N(2)-C(24)-C(23)-C(22)-C(21)-C(25)与苯环 C(25)-C(26)-C(21)-C(20)-C(19)-C(18)及吡啶环N(3)-C(15)-C(16)-C(17)-C(18)-C(26)部分重叠,环与环之间的质心距离为0.357 9 nm和0.369 7 nm,芳环的法线与质心矢量的夹角分别为1.94°和2.73°,说明分子间产生 π-π 堆积效应[17]。 这个 π-π堆积将配合物扩展为2D超分子结构(图4c)。分子中没有参与配位的游离DMF(N,N-二甲基甲酰胺)分子通过弱氢键C-H…O相互连接,镶嵌在2D超分子中,起着稳定分子结构的作用(图4d)。
2.2 配合物1~4的红外光谱
采用KBr压片法在4 000~400 cm-1范围内测定了配体和配合物的红外光谱,表4列出各物质的主要特征红外光谱数据。从表中可见,4个配合物中羧基的不对称伸缩振动和对称伸缩振动吸收峰均发生了不同程度的位移,这是因为羧基中的氧原子参与了配位,氧原子上的孤对电子被拉向金属离子,-C=O键力常数减小,造成观测到的羧基频率减小[18]。配合物2~4中-SO2的不对称伸缩振动和对称伸缩振动吸收峰基本不变,-SN键的振动吸收峰也基本不变,说明这2个基团的原子没有参与配位,而配合物1中-SO2的不对称伸缩振动吸收峰移动了14 cm-1。说明-SO2基团中的氧原子可能参与配位,晶体解析证实了这一点。但此配位键较弱,Cu-O键长为0.278 5(5)nm比正常的Cu-O配位键稍长些。
配体菲咯啉在 1 597、1 421、853 和 738 cm-1分别为C=N的伸缩振动峰,为C=C骨架振动,为C-H面外弯曲振动。形成配合物以后,配合物1分别移至 1 551、1 393、848和 723 cm-1; 配合物 2分别移至 1 552、1 393、848 和 726 cm-1;配合物 3 分别移至1 537、1 385、850 和 727 cm-1; 配合物 4 分别移至1 530、1 383、847 和 722 cm-1;其中 4 个配合物的C=N伸缩振动峰移动较多,其它吸收峰移动较少,说明菲咯啉的2个氮原子均与中心离子发生了配位[19]。
2.3 配合物1~4的热重差热分析
分析配合物的TG-DSC曲线(图5)可知:配合物1第一阶段失重出现在180~186℃,失重2.87%,约为1个配位水(理论值为2.14%),在186℃出现1个小的吸热峰;接着配合物一步分解完成,并在480℃处有1个很强的放热峰;到523℃趋于恒重,残重9.74%,估计残余物为CuO(理论值为9.50%)。配合物2是分步分解的,第一阶段失重出现在70~108℃,失重3.29%,约为1个结晶水分子 (理论值2.09%);第二阶段 108~145℃,失重 2.60%,约为 1个配位水分子(理论值2.09%);同时在108℃和145℃处出现2个小的吸热峰;145~265℃之间出现一平台,265℃后又开始失重,并出现1个很强的放热峰,到820℃时趋于恒重,最后残重9.50%,估计剩余产物为氧化锌(理论值9.39%)。配合物3的热稳定性较好,从255℃开始分步失重,到905℃时恒重,残重43.67%,估计剩余产物为PbO(理论值为46.07%)。配合物4也是分步分解,第一阶段失重出现在120~226℃,失重9.22%,约为1个DMF溶剂分子 (理论值为8.99%),到974℃趋于恒重,残重33.30%,估计残余物为PbO(理论值为27.46%),残余物比理论理值稍高些,可能是积碳的原因,分解的碳不能马上生成气体而夹在氧化铅中,导致残重含量高于理论值[20]。

表4 配体及配合物的主要红外光谱吸收峰Table 4 More relevant infrared bands for complexes 1~4 and ligand cm-1

图5 配合物1~4的TG-DSC曲线Fig.5 TG-DSC curves of the complexes 1(a),2(b),3(c)and 4(d)
2.4 配合物1~4的电化学性质
采用三电极体系,玻碳为工作电极、铂电极为辅助电极、SCE为参比电极,以DMF和水为溶剂,配合物的浓度为 1 μmol·L-1,2 mol·L-1KCl溶液为支持电解质。在氮气气氛中进行测定,从-1.0~1.2 V进行扫描,扫描速度为0.05 V·s-1。从循环图形来看,配合物1有1对氧化还原峰对应于电对Cu(Ⅱ)/Cu(Ⅰ)的电子转移,阴极峰和阳极峰的电位分别是Epc=-0.513 V,Epa=-0.337 V,从 ΔE=0.176 V 和 ipc/ipa=4.26,可以判别电解中电子转移是可逆的。配合物2有1对氧化还原峰对应于电对Zn(Ⅱ)/Zn(0)的电子转移,阴极峰和阳极峰的电位分别是Epc=-0.591 V和Epa=0.009 V,从ΔE=0.600 V和ipc/ipa=0.704,可以判别电解中电子转移是可逆的。配合物3只有1个还原峰,电位Epc=-0.955 V,ipc=0.180 μA,因此电子转移为不可逆。配合物4也只有1个还原峰,电位Epc=-0.609 V,ipc=0.180 μA,因此电子转移为不可逆。
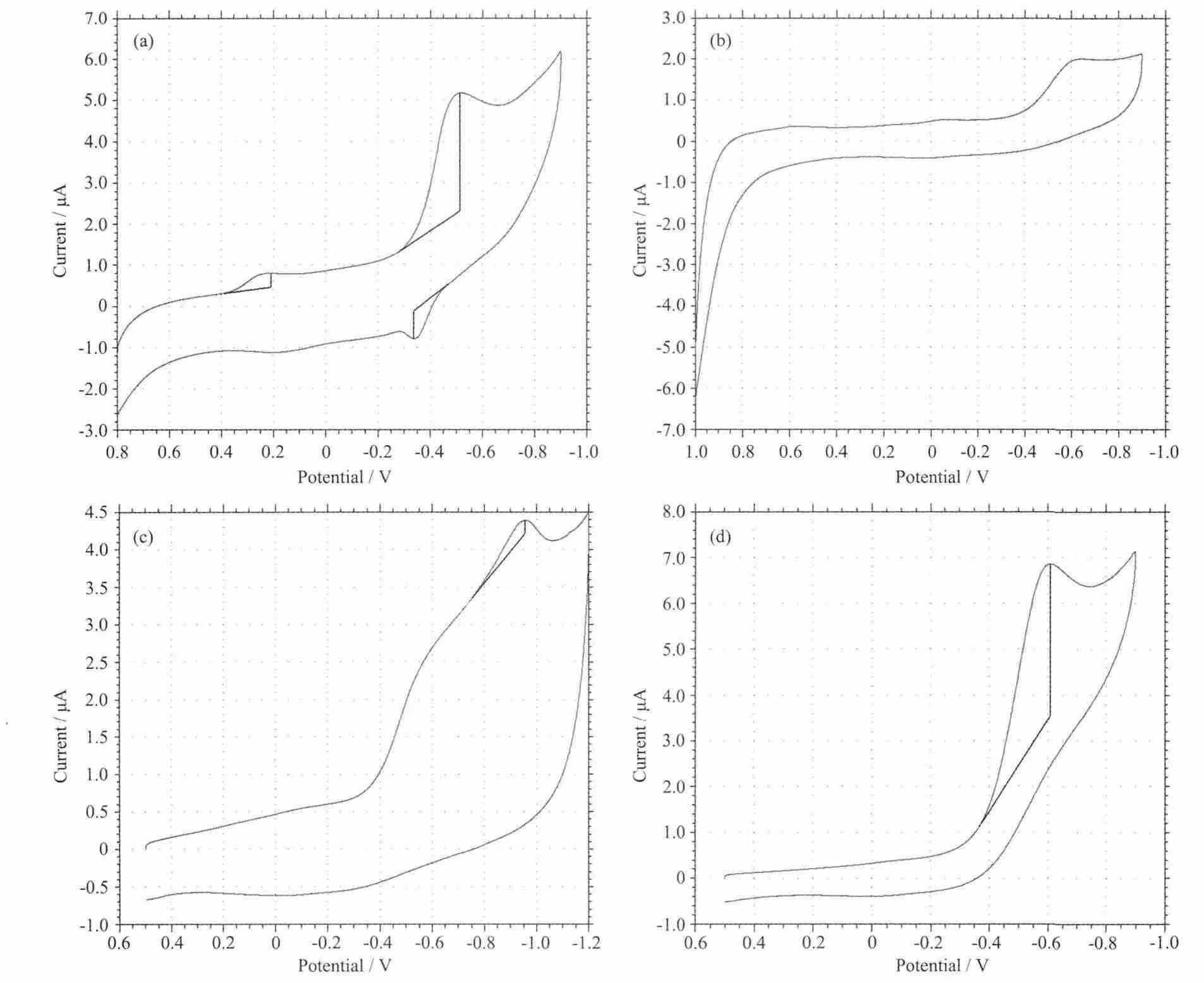
图6 配合物1~4的循环伏安图Fig.6 Cyclic voltammograms of the complexes 1(a),2(b),3(c)and 4(d)
2.5 配合物1~4的荧光性质
室温下,以N,N-二甲基甲酰胺为溶剂,配体和配合物浓度均为1×10-5mol·L-1,在荧光分光光度仪上测定了室温下配体和配合物在300~500 nm的发射光谱(见图 7)。

图7 配体和配合物2~4的发射光谱Fig.7 Fluorescence emission spectra of the ligand(a)and complexes 2(b),3(c)and 4(d)
由图7可见,配体Ts-p-aba在λem=336 nm处(λex=289 nm), 配合物 2 在 λem=336 nm 处 (λex=292 nm),配合物 3 在 λem=336 nm 处(λex=300 nm),配合物4在λem=336 nm处(λex=292 nm)均有荧光峰。配合物与配体Ts-p-aba的荧光光谱相似,说明配合物的荧光发射光谱主要来自配体Ts-p-aba的电荷跃迁,属LLCT发射(配体发射)。主配体Ts-p-aba的荧光峰较弱,因为配体分子中2个苯环不共面,共轭程度差[21]。生成配合物后,配合物2~4的荧光吸收强度均有较大的增加。这是因为金属离子配位增加了配体的电子共轭程度,减少了配体内部电荷转移带来的能量损失,且结构刚性增强的结果。3个配合物的荧光强度顺序为2<3<4,这与配合物的结构有关系,2为单分子,3为二聚体,4为聚合物,配合物中配体的刚性逐渐增强,且4中还存在着π-π堆积作用,因此荧光峰特别强。配合物1的荧光强度比配体还弱(在图中没有体现),可能是因为配体的荧光被铜离子猝灭的结果。
3 结 论
利用磺酰化对氨基苯甲酸及菲咯啉配体与3种不同金属离子自组装了4个配位化合物:[Cu(Tsp-aba)2(phen)(H2O)]n(1),[Zn(Ts-p-aba)2(phen)(H2O)]·H2O(2),[Pb(Ts-p-aba)2(phen)]2(3),{[Pb(Ts-p-aba)(phen)(NO3)](DMF)}n(4)。配合物1和4为一维直线链状结构,配合物2和3分别为零维单核分子和二聚体。中心离子的配位环境也不同,配合物1和3的中心离子Cu2+和Pb2+具有相似的八面体配位环境,而配合物2和4的中心离子Zn2+和Pb2+则分别具有四角锥和一面心八面体的配位环境。配体N-对甲苯磺酰对氨基苯甲酸在配合物1中采取双齿桥联的配位方式(其中磺酰基中的氧原子也参与配位,其它配合物中磺酰基的氧原子均没参与配位),2中采取单齿配位方式,3中采取双齿桥联/双齿螯合的配位方式,在4中则是螯合加单齿桥联。4个配合物组成和结构各不相同,导致热稳定性和电化学性质也各不同。随着聚合度增加配合物2、3、4的荧光强度也依次增大。这些结果说明,金属离子在此体系中起着导向作用,由于金属离子不同,导致配合物的晶体结构有很大的差异。从配合物3和4的结构来看,金属盐的阴离子对配合物的结构也有很大影响。
[1]O′Keeffe M,Yaghi O M.Chem.Rev.,2012,112:675-702
[2]Yang G P,Hou L,Luan X J,et al.Chem.Soc.Rev.,2012,41:6992-7000
[3]Cai S L,Zheng S R,Fan J,et al.J.Solid State Chem.,2011,184:3172-3178
[4]Deka H,Sarma R,Kumari S,et al.J.Solid State Chem.,2011,184:1726-1734
[5]Clemente-Leon M,Coronado E,Marti-Gastaldo C,et al.Chem.Soc.Rev.,2011,40:473-497
[6]Murray K S.Aust.J.Chem.,2009,62:1081-1101
[7]Du M,Wang Q,Wang Y,et al.J.Solid State Chem.,2006,179:3926-3936
[8]Liu S J,Zeng Y F,Hu X,et al.J.Solid State Chem.,2013,204:197-204
[9]WANG Shu-Ju(王淑菊),TIAN Yan-Wen(田彦文),YOU Li-Xin(由立新),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(3):511-516
[10]ZHANG Ting(张婷),CHEN Wen-Jian(陈文建),ZHANG Yun-Qian(张云黔),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(3):473-480
[11]Tong M L,Ye B H,Cai J W,et al.Inorg.Chem.,1998,37:2645-2650
[12]YANG Zi-Yan(杨滋渊),SONG Bin(宋斌),XU Shou-Wei(徐守卫),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(3):585-590
[13]Grirrane A,Pastor A,Galindo A,et al.Chem.Commun.,2003:512-515
[14]Beitone L,Huguenard C,Gansmuller A,et al.J.Am.Chem.Soc.,2003,125:9102-9110
[15]Robin A Y,Fromm K M.Coord.Chem.Rev.,2006,250:2127-2157
[16]Kitagawa S,Kawata S.Coord.Chem.Rev.,2002,224:11-34
[17]Ikai M,Ishikawa F,Aratani N,et al.Adv.Funct.Mater.,2006,16:515-519
[18]YU Shi-Lin(于世林),LI Yan-Wei(李寅蔚).Spectral Analysis(波谱分析法).Chongqing:Chongqing University Press,2000:44
[19]YANG Shu-Ping(杨树平),HAN Li-Jun(韩立军),PAN Yan(潘燕),et al.Acta Chim.Sinica(化学学报),2012,70(4):519-524
[20]ZHANG Rong-Nan(张荣兰),HU Juan(胡娟),ZHAO Jian-She(赵建社),et al.Acta Chim.Sinica(化学学报),2004,62(22):2259-2264
[21]XU Jin-Gou(许金钩),WANG Zun-Ben(王尊本).Fluorescence Analysis(荧光分析法).Beijing:Science Press,2006:21-22
