关联性特征在宏基因组分装中的应用*
2013-12-30张倩倩曹唱唱
张倩倩,曹唱唱,丁 啸,孙 啸
(东南大学生物科学与医学工程学院,南京210096)
宏基因组是指环境中全部微生物DNA的总和,最早是由Handelsman等人于98年在一篇研究土壤微生物的文章中提出[1]。宏基因组学,直接对混合的微生物群落样本进行基因组提取,然后以多种微生物基因组的混合体为测序模版,对其进行高通量测序。传统的研究方法是对单一微生物进行单纯培养,然后再对其进行分离测序研究,这样的方法停留在单一微生物物种的水平上。由于环境中99%的微生物都难以用常规的方法进行培养,所以传统的方法在很大程度上受到了限制。
然而,随着高通量测序技术的发展,当环境中的微生物样本被测序之后,需要对宏基因组测序片段建立种族进化关系[2],这样一个步骤叫做分装。分装也是一种特殊的聚类方式,指对宏基因组测序片段进行重叠区域保守拼接后根据一定的规则进行聚类,并将其归类到已知的系统发生关系中。归类的物种层次精度不一样,精确的可以归类至种,而粗糙的只能归类到界、门。归类的精度取决于多个因素如分装方法、群落结构和测序质量及深度[3]。微生物群落结构由研究样本所决定,测序质量和深度则依靠测序技术和样本尺寸。所以对于分装主要的研究重点就是分装方法。目前存在的分装方法分为2类:一类是基于序列相似性比对的分装方法;另一类是基于序列特征的分装方法。
基于序列相似性比对的分装方法其主要步骤是将宏基因组DNA片段进行处理之后与已知微生物基因组数据库做比对,然后再用比对所获得的信息对输入的测序片段进行分类和标注。最早的此类分装方法是由07年德国蒂宾根大学的Huson等人开发的MEGAN软件[4]。其原理直截了当,只需要用户利用序列比对工具BLAST将宏基因组测序数据与NCBI的微生物基因组数据库进行比对,生成一个软件可读取的比对中间文件。最后通过计算机分析,基于最近公共祖先算法将宏基因组DNA序列归类到物种分类树的相关结点上,并输出结果。但是这类算法太依赖数据库,局限性很大,毕竟我们已知的参考基因组数量有限,而且只有不到1%的微生物基因组可以培养和测序得到。于是研究者们开始分析并利用序列本身固有的成分特征来进行分装,从而提出了基于序列成分特征的分装方法。
基于序列特征的分装方法又包含2类:一类是基于物种分类标志物基因;另一类是基于碱基子串使用偏向性。物种分类标志物基因是指某一微生物在编码区域所特有的一段DNA序列,用于区分不同物种的标志,如传统的16S rRNA[5],以及新加入的rec A和rpo B基因等。利用这类特征进行分装具有以下局限性:首先,不是所有的宏基因组测序得到的DNA序列中都包含标志物基因的;其次,近年来科学家也发现,标志物基因也存在着种间平行转移的情况;最后,这种方法仍然还是依赖于物种分类标志物基因数据库,极大地限制了其应用范围。
针对以上限制,有学者提出使用基因组序列特征进行分装。目前主流的分装方法都是利用序列中碱基子串的组成特征,这种方法的理论依据是由Karlin等人在上世纪90年代中期对多种微生物全基因组序列进行相关研究后得出的[6-7]。他们发现同一物种基因组的不同片段有着非常相似的碱基子串使用偏向性,而不同基因组之间使用偏向性就很大。基于这样一种特性近年来也有很多新颖的分装方法,例如本文中用来做性能对比的由YANG等人提出的分装软件MetaCluster[8],还有Chan等人提出的基于自组织生长图(GSOM)算法[9-10]等。
基因组序列特征可分为序列组成性特征和关联性特征,而目前分装算法使用的都是组成性特征,如果能够在分装算法中引入关联性特征,则可望提高分装算法的性能。本文主要分析序列关联性特征在宏基因组分装上的应用。
1 基于序列关联性特征的分装方法
1.1 碱基对关联性
BBC特征反映的是碱基对的关联性(Base-Base Correlation)[11],该特征由序列的互信息 MIF(Mutual Information Function)定义而来。序列的互信息计算公式如下:
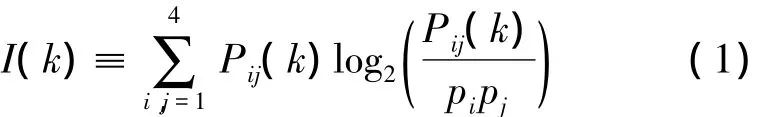
其中Pi表示单个核苷酸ni∈{A,G,C,T}出现的频率,Pij(k)表示一对被k个核苷酸分隔的核苷酸ni和nj出现的频率。这样I(k)表示,当识别到核苷酸X,得到相距k个核苷酸的核苷酸为Y时产生的信息量(以比特为单位)。举个例子来直观地说明I(k)的含义。
例子1 考虑一条随机序列,组成序列的各个核苷酸是独立无关的。直观上就可以看出,我们不能从X中得到任何Y的信息,所以对于任意k,I(k)都为零。事实上,从式(1)也可以得到相同的结果。由于所有的核苷酸统计上相互独立,所以由统计学公式可以得到:对所有的i、j和k,Pij(k)=PiPj。把这个式子代入到式(1)中,就可以得到对数中的因子为1,所以I(k)就为零。
由此我们可以定义序列的BBC特征如下:

其中Pi和Pij(l)的含义同上。Tij(k)表示不同间隔的二核苷酸组合在k+1长度上的平均相关性,反映了核苷酸序列的一种局部特征。
1.2 三联体核苷酸关联性
三联体核苷酸关联性特征,即序列中每一个三联体核苷酸单碱基与碱基对之间的关联性的量化。其定义为:宏基因组测序片段中三联体核苷酸第一位碱基与后两位二联体核苷酸的关联特性参数。这种特征一共包含了68个特征参数,分为2个部分。
第1部分是在确定第一位碱基的条件下后两位二联体核苷酸的出现频率,一共包含64个特征参数。这部分特征值的计算公式为:

其中nij表示确定第一位碱基后,三联体核苷酸的后两位核苷酸取16种不同碱基对时在序列中分别出现的次数表示确定第一位碱基的所有三联体核苷酸在序列中出现的次数。这部分特征值体现的是当第一位碱基确定之后,后两位核苷酸作为一个整体使用的频度。
第2部分是三联体核苷酸中的4种第一位碱基与后两位二联体核苷酸的相互信息量,一共包含4个特征参数。计算公式为:
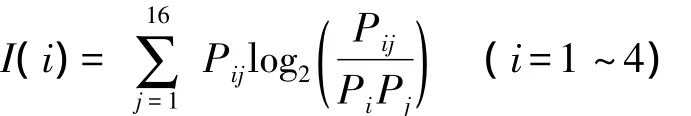
其中Pi表示第一位碱基在序列中出现4种类型的频率,Pj表示三联体核苷酸后两位核苷酸取特定碱基对时在序列中出现的频率,Pij表示指定三联体核苷酸在序列中出现的频率。1~4和1,2,…,16分别对应单碱基A、T、C、G和16种碱基对的编号。
1.3 机器学习算法
本文采用K均值分类法,K均值分类法原理是给定一个数据点集合和需要的聚类数目K,K均值算法根据某个距离函数反复把数据分入K个聚类中。其算法步骤为先随机选取K个对象作为初始的聚类中心。然后计算每个对象与各个种子聚类中心之间的距离,把每个对象分配给距离它最近的聚类中心。一旦全部对象都被分配了,每个聚类的聚类中心会根据聚类中现有的对象被重新计算。这个过程将不断重复直到满足某个终止条件。K均值分类法是一种无监督分类法,并以其简洁和效率而广泛使用。本文使用的K均值分类软件为Gene Cluster3.0[12]。
本文之所以选择K均值分类算法是因为首先这种算法分别是无监督方法,其次因为与作者提取的特征值进行对比的MetaCluster3.0软件里使用的分类算法就是K均值分类法,这样使用同一种分类法进行测试效果才有公平性。
2 实验结果及其讨论
2.1 模拟宏基因组测序数据集
从IMG[13]系统中根据分类树随机提取了18种微生物,其中包括2种古生菌、4种真核微生物、12种细菌。在此基础上根据不同的宏基因组数据复杂度创建了116组模拟数据集。首先根据分类学物种差异层次,数据集被分为同门异纲、同纲异目、同目异科、同科异属、同属异种5大组;然后我们在每个大组中,根据不同的基因组测序片段reads长度、测序错误率、相对丰度再进行模拟。当模拟数据集中的测序片段reads长度分别取500 bp、1 000 bp、2 000 bp、3 000 bp时,默认的测序错误率为1%,相对丰度为1∶1;当模拟数据集中的测序错误率分别取2%、3%、5%时,默认的 Reads长度为2 000 bp,相对丰度为1∶1;当模拟数据集中的相对丰度分别取1∶2、1 ∶4、1 ∶8时,默认的 Reads长度为 2 000 bp,测序错误率为1%。其中不同大组的数据集也会包含不同数量的微生物,其中可能有2种、3种、4种、8种或者10种微生物。
2.2 分类效果及其分析
对模拟数据集的分类结果分为2部分:第1部分是分别利用K均值分类法加入三联体核苷酸关联性特征参数和MetaCluster3.0软件对模拟数据集进行分类;第2部分是利用SVM分别加入三联体核苷酸关联性特征参数、三联体核苷酸使用频率和四联体核苷酸使用频率对模拟数据集进行分类。本文评估分类性能是根据分类准确率来判断,公式如下:

2.3 K均值分类效果对比及分析
由于模拟数据集比较多,限于篇幅作者只选取了一些代表性的数据集分类效果进行展示。选取的数据集和分类效果分别列在表1和表2。

表1 选取进行性能对比的模拟数据集
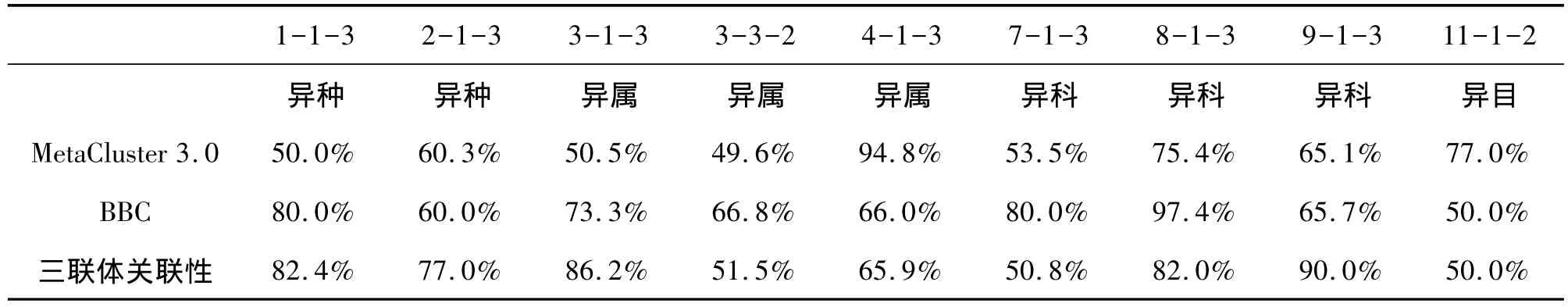
表2 MetaCluster3.0与碱基对关联性以及三联体核苷酸关联性方法的分装准确率对比
通过分析以上2张表格,我们可以发现在一样使用K均值分类方法时,MetaCluster3.0软件分类只有在同纲异目的物种层次时才能对模拟数据集进行较好的分装,在同科异属的包含3种微生物的数据集中表现出色,可能是由于数据集选择的微生物物种基因组中四联核苷酸使用频率恰好很相似的原因,因为作者通过对所有数据集分装后发现这是一个特例。通过数据也可以发现这款分装软件对宏基因组数据集的分类层次很敏感。反观三联体核苷酸关联性特征参数,分类效果多数情况下都要优于单纯使用四联核苷酸使用频率作为特征参数的Meta-Cluster3.0软件。
但是通过数据分析我们也发现,利用三联体核苷酸关联性特征参数进行分类存在两个问题:在对只包含真核微生物的数据集(数据集6-1-3,11-1-2)和丰度比不均匀的模拟数据集进行分类时效果不如意(数据集3-3-2)。作者认为可能因为真核微生物基因组较为复杂,其中包括大量非编码序列,而原核微生物基因组基本都是编码序列。而密码子就是由三位碱基组成,所以三联体核苷酸关联性特征参数对真核微生物的分类效果不好。对于丰度比不均匀导致分类效果不好,作者认为可能是由于使用的单纯的使用K均值分类法太过于简单,对丰度比比较敏感。
3 总结
得益于高通量测序技术的日渐成熟,宏基因组学在微生物群落的研究上进展迅速,但是同时伴随着一个问题。如何将大量的DNA短片段快速正确的分开,从而可以对宏基因组中的各种微生物进行研究,包括发现新物种、鉴定新基因及其发现微生物群的新功能等。宏基因组分装技术就是解决这个问题的核心,目前分装技术的主流是基于碱基使用偏向性特征的无监督算法。所以如何提取一种有效而又简单的序列特征参数是提升分装技术的重点。本文提出了一种三联体核苷酸关联性特征参数,它并不是单纯的使用三联体核苷酸使用频率,它体现的是序列中每一个三联体核苷酸单碱基与碱基对之间的关联性的量化。本文通过使用K均值分类法和支持向量机对模拟的不同复杂度的宏基因组数据集进行分类,发现利用三联体核苷酸关联性特征效果对大多数的数据集分类效果要优于无监督分装算法MetaCluster3.0,同时也好于单纯的利用三联、四联体核苷酸使用频率的分类效果。特别是在对种的分类中保持较高的准确率。但是这种特征值仍然存在问题,在使用无监督算法进行分类时,无法有效分类包含真核微生物和丰度比不均匀的模拟数据集。作者推测是由于三联体是密码子的结构,而真核微生物的基因组非编码序列要远远多于原核微生物基因组,导致三联核苷酸关联性特征失效。针对这个问题我们下一步准备基于四联核苷酸的关联性特征来设计分装算法。对于无法分类丰度比不均匀数据集的问题,我们准备尝试不同的机器学习算法或者对K均值分类法的距离进行改进,进一步的提高基于碱基关联性特征分装方法的准确性。
[1]Handelsman J,Rondon M R,Brady S F,et al.Molecular Biological Access to the Chemistry of Unknown Soil Microbes:A New Frontier for Natural Products[J].Chemistry and Biology,1998,5(10):R245-R249.
[2]McHardy A.Rigoutsos I.What’s in the Mix:Phylogenetic Classifcation of Metagenome Sequence Samples[J].Current Opinion in Microbiology 2007,10:499-503.
[3]Mavromatis K,Ivanova N,Barry K,et al.Use of Simulateddata Sets to Evaluate the Fidelity of Metagenomic Processing Methods[J].Nature Method,2007,4(6):495-500.
[4]Huson D H,Auch A F,Qi J,et al:MEGAN Analysis of Metagenomic Data[J].Genome Res,2007,17(3):377-386.
[5]Cole J R,Chai B,Farris R J,et al.The Ribosomal Database Project(RDP-Ⅱ):Sequences and Tools for High-Throughput rRNA A-nalysis[J].Nucleic Acids Res,2005,33(Database issue):D294-296.
[6]Karlin S,Ladunga I.Comparisons ofEukaryotic Genomic Sequences[J].Proc Natl Acad Sci USA,1994,91(26):12832-12836.
[7]Karlin S,Burge C.Dinucleotide Relative Abundance Extremes:A Genomic Signature[J].Trends Genet,1995,11(7):283-290.
[8]Yang B,Peng Y,Leung H C M,et al.Unsupervised Binning of Environmental GenomicFragmentsBased on an ErrorRobust Selection of l-Mers[J].BMC Bioinformatics,2010,11:1471.
[9]Chan C K,Hsu A L,Tang S L,et al.Using Growing Self-Organising Maps to Improve the Binning Process in Environmental Whole-Genome Shotgun Sequencing[J].J Biomed Biotechnol,2008,2008:513701.
[10]Chan C K,Hsu A L,Halgamuge S K,et al.Binning Sequences U-sing very Sparse Labels within a Metagenome[J].BMC Bioinformatics,2008,9:215.
[11]孙啸,傅静,焦典,等.生物信息学若干前沿问题的探讨:利用序列统计特征分析基因组序列[M].中国科学技术大学出版社,2004:51-56.
[12]Yang B,Peng Y,Leung H C M,et al.Unsupervised Binning of Environmental GenomicFragmentsBased on an ErrorRobust Selection of l-Mers[C]//BMC Bioinformatics 2010,11:1471.Liu Y Z,Moll J L,Spicer W E.Appl Phys Lett,1970,17(2):60-62.
[13]Markowitz V M,Chen I M A,Palaniappan K,et al.IMG:the Integrated Microbial Genomes Database and Comparative Analysis System[J].Nucleic Acids Research,2012,40:115-122.
