结直肠癌全基因组关联分析研究进展
2013-12-06来茂德朱益民综述
张 艳,来茂德,朱益民 综述
(浙江大学医学院1.流行病与卫生统计学系;2.病理学与病理生理学系,浙江杭州 310058)
结直肠癌(colorectal cancer,CRC)是最常见的恶性肿瘤之一,全球结直肠癌患病人数估计有280万,每年新增病例超过100万,发病率居于恶性肿瘤第三,死亡率处于第四位[1]。我国随着饮食结构和生活方式的改变,结直肠癌的发病率和死亡率呈不断上升趋势,2007年结直肠癌的发病率居恶性肿瘤第三位,死亡率居于第五[2]。结直肠癌已经严重威胁到居民的健康生活,并成为我国的社会负担。因此进一步研究结直肠癌的发病机制,降低结直肠癌的死亡率显得尤为重要。
结直肠癌是由环境和遗传因素共同起作用的结果,其中遗传因素在结直肠癌的发病中起重要的作用。研究发现结直肠癌在一级亲属的发病率是普通人群的2到3倍,基于双生子研究发现,大约35%结直肠癌的发生与遗传易感性有关[3]。经过多年的研究已发现了许多易感基因,然而,很多潜在的结直肠癌易感基因仍然没有被发现。近十年来,家系连锁分析已经发现了一些引起孟德尔疾病(如家族性腺瘤性息肉病)的罕见高危基因[4]。但是,连锁分析的关键是遗传因素完全或几乎完全决定疾病的发生,致病基因具有很高的外显率等。而结直肠癌作为复杂疾病被认为是受到多种基因的影响,每种基因的效应微弱,因此连锁分析在研究结直肠癌致病基因方面作用有限。比较大规模病例和对照人群之间某个基因型频率差异的关联研究被认为更适合用来识别复杂疾病(包括结直肠癌)的易感位点。全基因组关联研究(genome-wide association studies,GWAS)是利用高通量的基因芯片技术,主要对个体中数以百万计的单核苷酸多态性(SNPs)进行检测研究,在全基因组的水平上、在大样本人群中进行病例对照关联分析,发现与疾病相关的阳性位点,然后将此阳性位点在独立的样本中进行验证,从而发现影响复杂性疾病发生的遗传易感变异[5]。迄今为止,利用GWAS已经发现了一些与结直肠癌相关联的疾病易感位点和区域,为进一步研究结直肠癌的遗传机制,有必要对结直肠癌GWAS研究进展作一综述。
1 结直肠癌GWAS现状
从2007年开始许多国家对结直肠癌开展了多项GWAS,在结直肠癌遗传学研究方面取得了一定的进展,发现了一些与之相关的易感位点与区域(表1)。
1.1 欧美人群的结直肠癌GWAS
1.1.1 结直肠癌遗传性(colorectal cancer genetics,COGENT)研究 COGENT研究小组在英格兰、苏格兰、加拿大人群中通过多中心大规模样本的重复验证,先后发现了11个结直肠癌的易感位点:rs6983267(8q24)[6]、rs10505477(8q24)[7]、rs719725(9p24)[7]、rs4939827(SMAD7)[8,11]、 rs4779584 (GREM1)[9]、rs16892766 (EIF3H )[10]、 rs10795668(10p14)[10]、rs3802842(11q23)[11]、rs7014346(8q24)[11]、rs1957636(BMP4)[16]、rs4813802(BMP2)[16]。这些位点效应都较弱,OR 值大部分在1.10~1.30之间。为了发现效应更小的易感位点,对英国人群的GWAS数据进行Meta分析,以及病例对照的验证,发现了另外8个新的易感位点:rs10411210(RHPN2)、rs4444235(BMP4)、 rs961253(BMP2)、rs9929218(CDH1)[12]、rs10936599(3q26.2)、rs11169552(12q13.13)、rs4925386(20q13.33)、rs6691170(1q41)[14]。然而,这些位点效应更弱,OR值在1.10左右。
1.1.2 欧洲其他人群研究 Lascorz[13]等2010年对德国371家族性结直肠癌病例和1263例对照进行GWAS分析,经过4个独立的病例对照研究验证,新发现了rs12701937(GLI3/INHBA)在显性模型中与结直肠癌的发病风险存在关联(OR=1.14),且此关联在家族性结直肠癌病例中的效应更强(OR=1.36)。另外,GWAS筛选出的不同位点已成功地在不同欧美人群中得到验证,如rs16892766[17]、rs6983267[18-22]、rs10505477[7]、rs3802842[17,21,23]、rs4444235[24]、rs4779584[21]和 rs4939827[18,25]。
1.2 亚洲人群的结直肠癌GWAS 2011年,Cui[15]等首次报道了在亚洲人群的结直肠癌GWAS结果。该小组在6q26~q27区域发现了新位点,SLC22A3基因上的rs7758229与远端结肠癌的发病风险存在显著关联(P=7.92×10-9)。该研究结果提示,在欧洲和亚洲人群之间结直肠的发病机制存在一定的种族差异。
1.3 结直肠癌易感位点在非洲黑人人群的重复验证研究 结直肠癌的GWAS在非洲人群中尚未进行,但是He[21]等在一个多种人群中对11个GWAS发现的结直肠癌易感位点进行重复验证研究,结果发现,在美国黑人中rs6983267(8q24)和rs961253(20p12)与结直肠癌/腺癌存在显著关联。
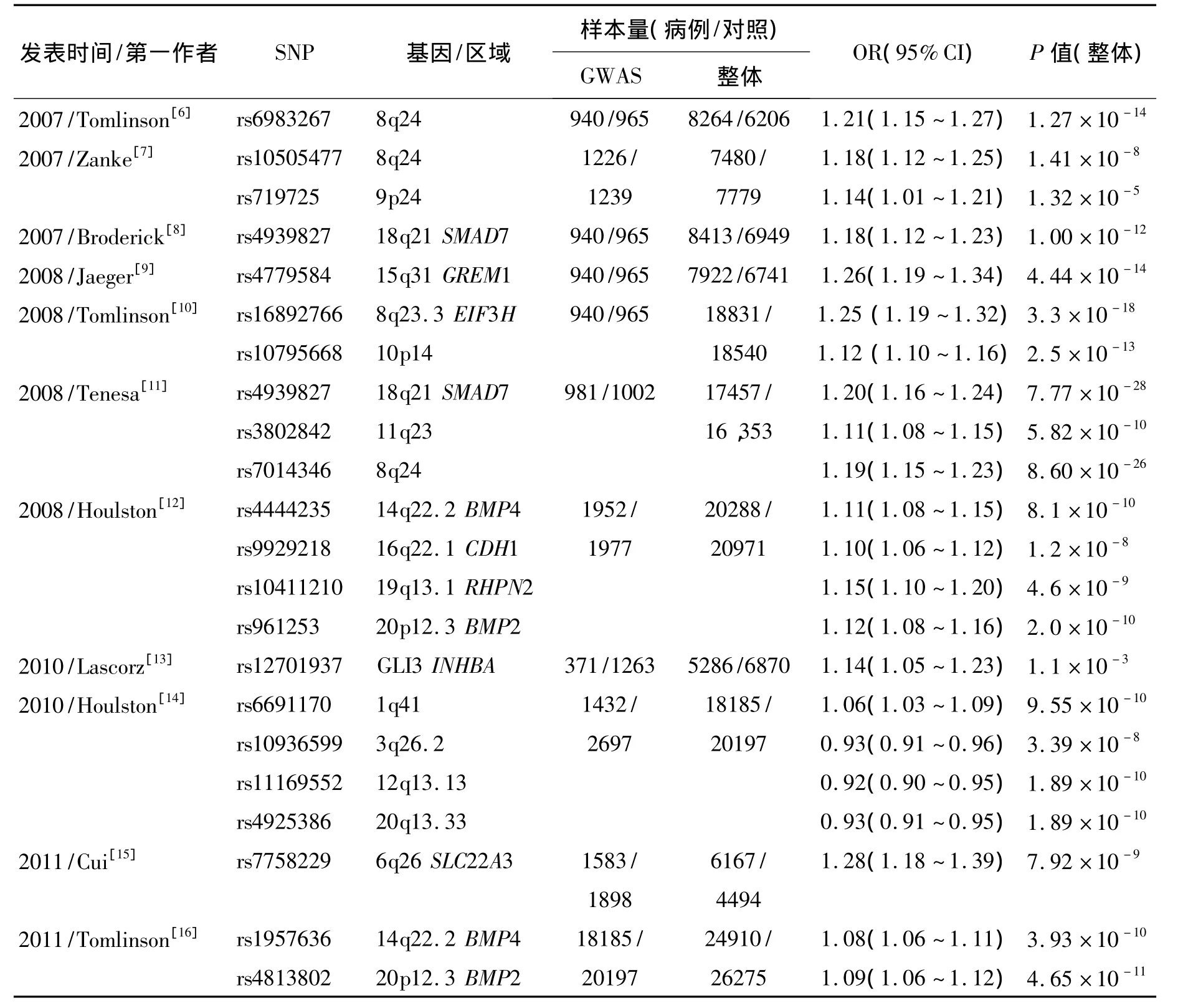
表1 结直肠癌GWAS筛选出的SNP标志Table 1 Loci associated with colorectal cancer risk from GWAS
总之,GWAS发现的与结直肠癌有关的易感位点效应都较弱,OR值大部分都在1.10~1.20之间。所有位点均位于非编码内含子区域,一些缺乏编码序列或是转录活性的区域称为基因沙漠[26]。比如最早发现与结直肠癌有关的易感位点rs6983267[6-7]位于距离最近的基因330 kb之外的基因沙漠区。另外,在已报道的易感位点中有5个位点标记的连锁不平衡区块包含或邻近转化生长因子-β(TGF-β)超家族信号通路中的基因,它们是SMAD7[12,16]、GREM1[9]、骨形成蛋白 BMP2 和 BMP4 以及RHPN2[14,20]。众所周知,TGF-β 超家族蛋白与细胞的增殖、分化和迁移有关,提示TGF-β超家族在结直肠癌中有一定的作用[27],这也说明虽然这些易感位点对结直肠癌的效应较小,但是它们的功能效应可能很大。还有3个易感位点位于或邻近某基因,分别为 rs7758229(SLC22A3)、rs16892766(EIF3H)和 rs9929218(CDH1)。SLC22A3是有机阳离子转运家族的一员,该家族在转运阳离子药物、毒物和内源性代谢物中起关键的作用[15],而许多毒物或内源性代谢物能引起肿瘤的形成;EIF3H调节细胞的生长和发育[10],然而在 1q41、3q26.2、8q24、9p24、10p14、11q23、12q13.13 和 20q13.33 上的位点都位于没有已知生物学关联的基质间隔区,所在区域都没有明显的候选基因。因此,仍然需要大量的研究探寻这些关联的生物学机制。
2 结直肠癌GWAS的优势与不足
2.1 结直肠癌GWAS的优点 GWAS是一种具有高通量、高保真度、无假设的分析方法,对于结直肠癌这类复杂疾病的研究中,具有很大的优势。之前的研究已经发现一些已知基因的高外显性,种系突变只能用于解释小于5%的结直肠癌病例[28],剩余的遗传变异则归功于大量常见的、遗传效应很弱的易感位点。相较于候选基因法和连锁分析,GWAS通过对全基因组进行扫描比较大规模病例和对照人群之间某个基因型频率差异,不需要先有候选基因,再分析基因与遗传标志的变异,可以发现大量的常见变异,以及一些对结直肠癌遗传效应极小的遗传变异,甚至能发现未知的基因。另外,大样本量的采用、多阶段的重复验证以及强大的统计分析方法极大地提高了检出结直肠癌相关易感位点的效能。
另外,相对于一般GWAS选择少量的峰值位点进行后期验证,虽然降低了假阳性率,但也掩盖了许多其他易感位点,提高了假阴性率。目前几例结直肠癌GWAS为了发现更多的易感位点,通过扩大验证位点的数量,降低检验水平等手段,取得了不错的成效[10-11]。
2.2 GWAS自身的局限性 同其他方法一样,GWAS不可避免地会存在一些不足,由于GWAS自身的局限性,使得在结直肠癌遗传易感性的研究中存在了一些问题。我们要正视GWAS存在的问题,采取合理的措施尽可能弥补这些不足,进一步优化GWAS和后续的研究,发现更多的与结直肠癌致病机制相关的易感位点。
2.2.1 GWAS检出的易感位点功能多不明确
尽管GWAS发现了许多与结直肠癌相关的易感位点,但是所有位点均位于非编码区,有的甚至远离基因编码区,即使有的易感位点所在的连锁不平衡区块包含或邻近某基因,但是这些基因对结直肠癌的具体功能也多不明确。因此需对这些区域进行重测序、精细作图寻找其他关联或致病位点,并对关联或致病位点进行体内外的功能验证,最终把GWAS的研究结果与生物学研究联系起来。
2.2.2 GWAS对低频 SNPs检出效能不够
GWAS发现的结直肠癌相关变异均是常见变异(MAF平均值在0.39左右)。低频变异(MAF<0.05)很难被检出,针对这个问题,增加GWAS的样本量是最直接的方法,但鉴于GWAS的巨大花费,现有的样本量难以识别出这些低频SNPs。Meta分析通过合并多个研究数据增加样本量,提高统计效能,不仅有利于发现新的常见变异,也能发现一些遗传效应强的罕见变异[29]。通过充分利用一些信息,如数量性状位点的表达分析等,候选基因法渐渐被认为是一种可以发现低频变异的方法[16]。特定区域的重测序和精细作图以及全基因组测序研究也可能发现一些罕见变异[30]。
2.2.3 GWAS对遗传变异的研究主要集中在SNPs 目前结直肠癌GWAS研究的主要对象为SNPs,对于其他的变异研究较少,如拷贝数变异(copy number variations,CNVs),小片段的缺失,串联重复序列和其他结构的变异等。有研究发现,位于3q26的CNV区域可能与APC基因突变阴性的家族性结直肠癌发病有关[31],这也提示CNVs可能是另一结直肠癌的易感变异。一项基因表达变异研究发现,SNPs和CNVs分别与83%和18%的基因表达变异有关[32],相较于 SNPs我们对 CNVs的了解还很不全面,有可能低估CNVs的作用。随着千人基因组计划的推进,有望使人类基因组CNVs的图谱更加清晰;随着检测分析技术的不断发展,有望为阐明CNVs在结直肠癌遗传机制中的作用提供平台[33],在结直肠癌GWAS中加入CNVs的检测,有利于进一步解释结直肠的遗传易感机制。
2.2.4 GWAS 忽略或淡化了基因-基因和基因-环境的交互作用 目前结直肠癌GWAS更多关注的仍是遗传因素对结直肠癌的影响,许多研究者简化了结直肠癌的发病机制的复杂性,把此主要归结为多个易感位点或基因的影响,对基因-基因、基因-环境联合作用研究的较少。将不同的暴露人群混合后寻找遗传易感位点,由于暴露的混杂效应会削弱发现关联的能力,降低研究效能,从而导致大量的遗传变异被掩盖[34]。在一个移民及时间趋势的研究中显示环境因素对结直肠癌的发病有很强的作用[35]。所以在研究遗传与结直肠癌的关联时,应关注遗传-环境之间的交互作用。另外可以采用全基因组单倍体分析研究基因-基因作用与结直肠癌的关系。
2.3 结直肠癌GWAS的不足 在研究结直肠癌遗传易感性的过程中,除了GWAS自身的局限性造成的问题外,针对结直肠癌这一特定病种还存在其他的不足之处。目前结直肠癌GWAS主要集中在欧美人群,只有一例在亚洲人群中进行,而且与结直肠癌相关的易感位点在其他种族的重复验证研究也较少。由于不同种族间结直肠癌的发病率、遗传变异的频率、连锁不平衡的模式存在很大的差别,因此与结直肠癌相关的易感位点可能也会不同。比如在亚洲人群中[15]发现SLC22A3基因上的rs7758229与远端结肠癌的发病风险存在显著关联,而该位点在此前多个欧美人群中均未报道。另外GWAS发现的一些结直肠癌易感位点存在部位特异性:rs3802842(11q23.1)[11]、rs4939827(18q21.1)[11,18]和 rs6691170(1q41)[36]与直肠癌的发病风险有关,而与结肠癌的发病风险增加无关;在亚洲的 GWAS[15]中发现,SLC22A3基因上的rs7758229只与远端结肠癌的发病风险存在显著关联。提示遗传因素对直肠癌、远端结肠癌、近端结肠癌的发病机制贡献不同。因此在研究结直肠癌遗传变异的过程中部位特异性的研究也是非常必要的,而目前的GWAS中相关的数据则很有限。针对这些问题,迫切需要更多亚洲和非洲人群的结直肠癌GWAS数据,以及针对肿瘤不同部位、类型的研究。
3 结直肠癌GWAS的应用与展望
3.1 结直肠癌GWAS的应用
3.1.1 结直肠癌GWAS与公共卫生 单个位点的检测对结直肠癌的预测贡献很小,携带遗传易感位点个体与未携带者相比患结直肠癌的风险也只是略有不同。但是通过GWAS对全基因组进行扫描,识别一连串与结直肠癌相关的易感位点,形成结直肠癌遗传易感谱,通过检测多个易感位点,根据多个位点的数据建立一个风险预测模型,对结直肠癌进行预测分析,从而将易感人群从一般人群中筛选出来,这对结直肠癌的防治有积极的作用[34]。但是必须指出的是遗传因素只是发病机制中一个重要的部分,并不是引起疾病的唯一原因,所以用于疾病预测的观点目前只是属于研究阶段,并没有想象的简单,要真正地上升到公共健康这一层面还需要很大的努力。
3.1.2 结直肠癌GWAS与临床应用 结直肠癌患者中携带的易感位点不同,其肿瘤类型也可能不同,Lubbe等[36]在3146名结肠癌患者中对易感位点与肿瘤分型进行关联分析,结果发现一些位点与特定的肿瘤类型显著相关,临床上可根据患者易感位点不同采用不同的治疗方法。Xing[37]等对GWAS识别出的易感基因进行了疾病预后的关联研究,得出10p14上的rs10795668与结直肠癌的复发风险率降低有关(HR=0.55,P=0.05),并提示该位点可能是作为识别化疗后复发的标志物;另外,15q31上的rs4779584则与结直肠癌的生存率有关,可用于疾病预后研究。
3.2 结直肠癌GWAS的展望 尽管GWAS发现的结直肠癌的易感位点和区域只是冰山一角,对于阐明结直肠癌的遗传特性的作用似乎微乎其微,大部分发现的SNPs只能轻度地增加结直肠癌的风险[38],但是结直肠癌GWAS的发现能够促进基因组学中一些基础研究(如功能学研究)的进行,进一步了解结直肠癌的易感机制,探讨结直肠癌的发病机制。随着技术的进步,GWAS的优化,在未来可能会发现更多的新的基因、位点、通路,结合后续的功能研究,从而发现真正的结直肠癌的致病基因,把研究结果与实际应用相结合,进而降低结直肠癌的人群患病率和死亡率。
[1]BOYLE P,LEVIN B.World cancer report 2008[M].Lyon:International Agency for Research on Cancer(IARC),2008:380-384.
[2]CHEN Wanqing,ZHANG Siwei,ZHENG Rongshou,et al(陈万青,张思维,郑荣寿,等).A report of cancer incidence and mortality from 38 cancer registries in China,2007 [J].China Cancer(中国肿瘤),2011,20(3):162-169.(in Chinese)
[3]LICHTENSTEIN P,HOLM N V,VERKASALO P K,et al.Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden,Denmark,and Finland [J].New England Journal of Medicine,2000,343(2):78-85.
[4]LE MARCHAND L.Genome-wide association studiesand colorectalcancer [J].Surgical Oncology Clinics of North America,2009,18(4):663-668.
[5]HARDY J,SINGLETON A.Genomewide association studies and human disease [J].New England Journal of Medicine,2009,360(17):1759-1768.
[6]TOMLINSON I,WEBB E,CARVAJALCARMONA L,et al.A genome-wide association scan of tag SNPs identifiesa susceptibilityvariantforcolorectal cancer at 8q24.21 [J].Nature Genetics,2007,39(8):984-988.
[7]ZANKE B W,GREENWOOD C M T,RANGREJ J,et al.Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24 [J].Nature Genetics,2007,39(8):989-994.
[8]BRODERICK P, CARVAJALCARMONA L,PITTMAN A M,et al.A genome-wide association study showsthatcommon allelesofSMAD7 influence colorectalcancerrisk [J].Nature Genetics,2007,39(11):1315-1317.
[9]JAEGER E,WEBBE,HOWARTHK,etal.Common genetic variants at the CRAC1(HMPS)locus on chromosome 15q13.3 influence colorectal cancer risk [J].Nature Genetics,2008,40(1):26-28.
[10]TOMLINSON I P M, WEBB E,CARVAJALCARMONA L,et al.A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3 [J].Nature Genetics,2008,40(5):623-630.
[11]TENESA A,FARRINGTON S M,PRENDERGAST J G D,etal.Genome-wideassociationscan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21[J].Nature Genetics,2008,40(5):631-637.
[12]HOULSTON R S,WEBB E,BRODERICK P,et al.Meta-analysis ofgenome-wide association data identifies four new susceptibility loci for colorectal cancer[J].Nature Genetics,2008,40(12):1426-1435.
[13]LASCORZ J,FORSTI A,CHEN B,et al.Genomewide association study for colorectal cancer identifies risk polymorphisms in German familial cases and implicates MAPK signalling pathways in disease susceptibility[J].Carcinogenesis,2010,31(9):1612-1619.
[14]HOULSTON R S,CHEADLE J,DOBBINS S E,et al.Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41,3q26.2,12q13.13 and 20q13.33[J].Nature Genetics,2010,42(11):973-978.
[15]CUI R,OKADA Y,JAGN S G,et al.Common variant in 6q26-q27 is associated with distal colon cancer in an Asian population [J].Gut,2011,60(6):799-805.
[16]TOMLINSON I P M,CARVAJALCARMONA L,DOBBINS S E, et al.Multiple Common Susceptibility Variants near BMP Pathway Loci GREM1,BMP4,and BMP2 explain part of the missing heritability of colorectal cancer[J].Plos Genetics,2011,7(6):1-11.
[17]WIJNEN J T,BROHET R M,VAN EIJK R,et al.Chromosome 8q23.3 and 11q23.1 variants modify colorectal cancer risk in Lynch Syndrome[J].Gastroenterology,2009,136(1):131-137.
[18]CURTIN K,WEI Y L,GEORGE R,et al.Meta association ofcolorectalcancerconfirmsrisk allelesat8q24 and 18q21 [J].Cancer Epidemiology Biomarkers& Prevention,2009,18(2):616-621.
[19]YEAGER M,XIAO N Q,HAYES R B,et al.Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers [J].Human Genetics,2008,124(2):161-170.
[20]SCHAFMAYER C,BUCH S,VOELZKE H,et al.Investigation of the colorectal cancer susceptibility region on chromosome 8q24.21 in a large German case-control sample [J].International Journal of Cancer,2009,124(1):75-80.
[21]HE J,WILKENS L R,STRAM D O,et al.Generalizability and epidemiologic characterization of eleven colorectal cancer GWAS hits in multiple populations [J].Cancer Epidemiology Biomarkers & Prevention,2011,20(1):70-81.
[22]HAERIAN M S,BAUM L,HAERIAN B S.Association of 8q24.21 loci with the risk of colorectal cancer:a systematic review and metaanalysis[J].Journal of Gastroenterology and Hepatology,2011,26(10):1475-1484.
[23]PITTMAN A M,WEBB E,CARVAJALCARMONA L,et al.Refinement of the basis and impact of common 11q23.1 variation to the risk of developing colorectal cancer [J].Human Molecular Genetics,2008,17(23):3720-3727.
[24]LIJ,SUN C,YUAN Y R,etal.Bone morphogenetic protein-4 polymorphism and colorectal cancer risk:a meta analysis [J].Molecular Biology Reports,2012,39(5):5239-5251.
[25]PITTMAN A M,NARANJO S,WEBB E,et al.The colorectal cancer risk at 18q21 is caused by a novel variant altering SMAD7 expression [J].Genome Research,2009,19(6):987-993.
[26]GOEL A,BOLAND C R.Recent insights into the pathogenesis of colorectal cancer[J].Current Opinion in Gastroenterology,2010,26(1):47-52.
[27]TENESA A,DUNLOP M G.New insights into the aetiology of colorectal cancer from genome-wide association studies [J].Nature Reviews Genetics,2009,10(6):353-358.
[28]AALTONEN L,JOHNS L,JAERVINEN H,et al.Explaining the familialcolorectalcancerrisk associated with mismatch repair(MMR)-deficient and MMR-stable tumors[J].Clinical Cancer Research,2007,13(1):356-361.
[29]QUAN Sheng,ZHANG Xuejun(权 晟,张学军).Research strategies for the next step of genome-wide association study[J].Hereditas(遗传),2011,33(2):100-108.(in Chinese)
[30]CIRULLI E T,GOLDSTEIN D B.Uncovering the roles of rare variants in common disease through whole-genome sequencing [J].Nat Rev Genet,2010,11(6):415-425.
[31]THEAN L F,LOI C,HO K S,et al.Genome-wide scan identifies a copy number variable region at 3q26 that regulates PPM1L in APC mutationnegative familial colorectal cancer patients[J].Genes Chromosomes Cancer,2010,49(2):99-106.
[32]STRANGER B E,FORREST M S,DUNNING M,et al.Relative impact of nucleotide and copy number variation on gene expression phenotypes[J].Science,2007,315(5813):848-853.
[33]XI Bo,MI Jie(席 波,米 杰).Study on genome wide association in patients with human obesity[J].Chin J Epidemiol(中华流行病学杂志),2010,31(12):1425-1428.(in Chinese)
[34]YANG Ying,LU Xiangfeng(杨 英,鲁向锋).Advances in genome-wide association study of coronary heart disease[J].Hereditas(遗传),2010,32(2):97-104.(in Chinese)
[35]LE MARCHANDL,WILKENSLR.Design considerations for genomic association studies:Importance of gene-environment interactions[J].Cancer Epidemiology Biomarkers &Prevention,2008,17(2):263-267.
[36]LUBBE S J,WHIFFIN N,CHANDLER L,et al.Relationship between 16 susceptibility loci and colorectal cancer phenotype in 3146 patients[J].Carcinogenesis,2012,33(1):108-112.
[37]XING J,MYERS R E,HE X,et al.GWAS-identified colorectal cancer susceptibility locus associates with disease prognosis[J].European Journal of Cancer,2011,47(11):1699-1707.
[38]TU Xin,SHI Lisong,WANG Fan,et al(涂 欣,石立松,汪 樊,等).Genomewide association study:advances,challenges and deliberation [J].Progress in Physiological Sciences(生理科学进展),2010,41(2):87-94.(in Chinese)
