高通量测序技术分析肺结核患者PBMC基因表达谱差异①
2013-11-28张晨晨张国良张明霞陈心春孟志忠
魏 晶 张晨晨 张国良 张明霞 陈心春 孟志忠
(华南理工大学生物科学与工程学院,广州510006)
据世界卫生组织(WHO)统计,全球约有1/3的 人感染了结核杆菌,每年新发感染人数约800万,每年因结核病而死亡的人数更高达200万[1]。我国结核杆菌的感染率为44.5%,活动性肺结核患者451万,每年死亡人数达13万,加上目前人类免疫缺陷病毒(HIV)感染的流行,结核杆菌耐药菌株的出现,导致结核病疫情急剧恶化,并且机体在感染肺结核后,对病原体免疫的控制和防护的具体途径和机制知之甚少[2,3]。人类对于疾病的认识最终是建立在对基因组及其功能的全面理解的基础上[4],新一代高通量测序技术有助于从基因水平对疾病进行研究,并在各个领域如癌症、HIV、肝病等都取得了较好效果[5-8]。因此本研究采用Illumina高通量测序技术对肺结核患者的mRNA表达谱进行分析,以期找到用于肺结核诊断的特异性标志物。
1 材料与方法
1.1 病人和标本收集 参照中国卫生部《结核病临床诊疗指南》,在深圳市第三人民医院收集10例肺结核患者[男女各5例,年龄(22.1±5.13)岁]的全血20 ml,所有进行结核菌特异性IFN-Υ ELISPOT检测斑点数>30个,无合并慢性疾病与自身免疫疾病[9]。健康对照组10例[男女各5例,年龄(23.7±5.10)岁]均来自深圳市第三人民医院体检人员。
1.2 外周血单个核细胞(PBMC)的分离和RNA的提取 本研究采用了Ficoll-hypaque(葡聚糖-泛影葡胺)密度梯度离心法分离全血中的PBMC(确保细胞浓度为106ml-1),并用Trizol法提取PBMC中的RNA,用NanoDrop 2000分光光度计测总RNA的浓度/纯度,琼脂糖凝胶变性电泳检测RNA的完整性。
1.3 cDNA文库的构建和确立及Illumina高通量测序 等量混合10例肺结核患者RNA组成RNA池,健康人群RNA也采取同样的处理方法,按照标准的Illumina 1.5说明书构建cDNA文库。首先用Poly(T)寡聚核苷酸从总RNA中抽取全部带Poly(A)尾的mRNA,将所得的mRNA随机打断成片段,再用随机引物和逆转录酶从mRNA片段合成cDNA片段。然后,对cDNA片段进行末端修复并连接测序接头,得到将用于测序的cDNA。为了提高测序效率,本实验还用电泳切胶法获取长度范围在200 bp(±25 bp)的cDNA片段,再通过 PCR扩增,得到最终的cDNA文库。将所得的cDNA文库加入流动槽中的各通道中,经过桥式 PCR扩增后,用Illumina Genome Analyzer IIx测序。
1.4 高通量测序数据的标签(tag)分析,基因表达量的标准化 测序后得到的是带有3'接头的原始序列数据,含有各种杂质以及少量低质量标签,去除一系列包含N和拷贝数小于2的标签后,得到高质量标签(Clean_Tag)。将Clean_Tag与人类参考基因hg18比对,取错配数小于2并唯一比对的基因作为进一步分析的基因。为了准确、科学地衡量每个基因的表达水平,对每个基因表达量做标准化处理,标准化结果用TPM(transcript per million clean tags)表示[10]。
1.5 差异表达基因的筛选 参照文献[11]报道的方法,采用严格的算法筛选两样本间差异表达的基因,并对差异检验的P值进行多重假设检验校正,然后通过控制FDR(false discovery rate)假阳性率来最终决定P的阈值[12]。本研究差异表达基因定义为“FDR≤0.001”且差异倍数2倍以上(即 |log2 Ratio|≥1)的基因。
1.6 Go功能富集分析与KEGG Pathway显著性富集分析 对于差异表达的基因进行基因本体(gene ontology,GO)分析,确定差异表达基因的功能,对P值矫正,满足矫正P值≤0.05的GO条目被认为是显著性富集的条目。对差异基因进行KEGG Pathway分析,其目的在于确定不同样本间差异表达的基因所参与的最主要代谢途径和信号转导途径。
2 结果
2.1 整体分析高通量测序数据 为了解肺结核患者体内基因表达的情况,我们构建了2个cDNA文库:肺结核PBMC文库和正常对照组PBMC文库,然后将其用Illumina GAIIx高通量测序技术测序。对于肺结核样本,测序后产生的原始标签(raw tags为3839500,对应的标签种数(distinct tags)为293077类,过滤掉拷贝数小于2和包含N的tag,还剩下3651827个107200类高质量标签(clean_tag),将clean_tag与人类参考基因组的明确的tag比较,只取唯一定位到参考序列,并且错配数小于2的tag,最后得到2430312个36390类tag,基因数为12270个。我们所用的人类参考基因为hg18(NCBI Build 36.1),其基因数30456,明确的 tag数 275988,占总tag数的92.61%。正常对照组最后产生了12244个基因,其他的数据如表1所示。
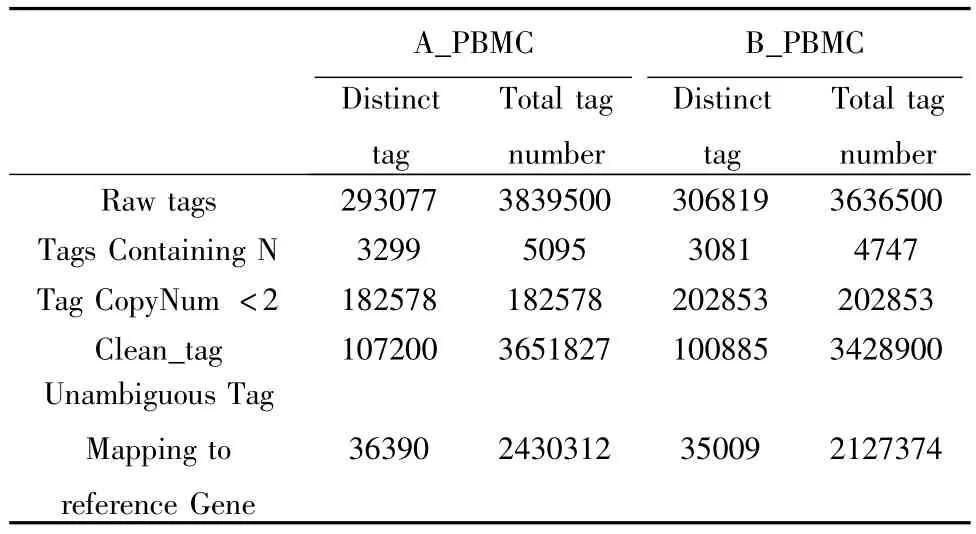
表1 肺结核和正常对照样本中序列标签分布一览表Tab.1 The distribution list of Tuberculosis and health control samples sequencing tags

图1 肺结核PBMC与正常对照PBMC基因表达水平的比较Fig.1 Gene expression level A_PBMC vs B_PBMC
2.2 差异基因的筛选 将唯一比对上人类参考序列的基因表达量标准化后筛选肺结核和正常对照差异表达的基因(图1)。取A_PBMC/B_PBMC比值的log2值,并筛选倍数≤-1(指在A_PBMC样本中表达量低),倍数≥1(指在A_PBMC样本中表达量低),FDR<0.001的基因,最终筛选出了3 097个基因,其中A_PBMC>B_PBMC(上调)的基因1 601个,A_PBMC<B_PBMC(下调)的基因1 469个。将log2 Ratio值进一步增大筛选标准至4倍,上调的基因数为74个,下调的基因数为269个。再对上调的基因控制A_PBMC的表达量>20,筛选出了16个差异极其显著的基因,鉴于下调的基因数较多并且B_PBMC的表达量范围分布较广(图2),因此筛选了17个表达量大于300的基因作为差异极显著的基因(表2)
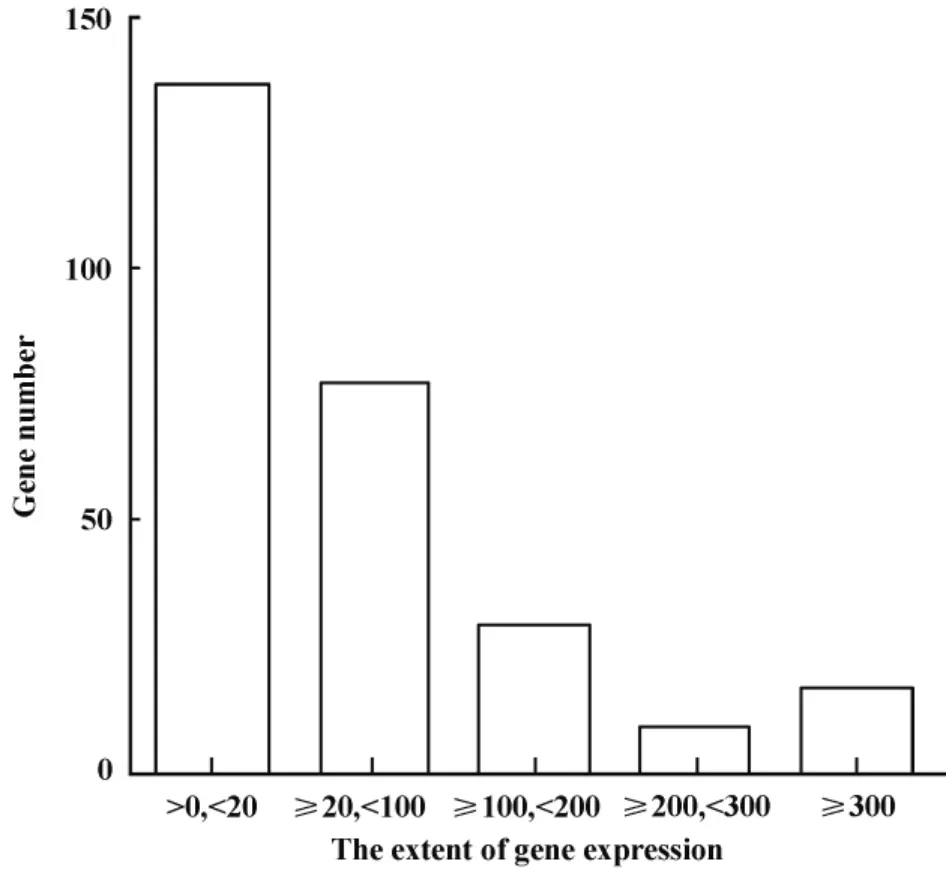
图2 269个下调基因的表达量分布Fig.2 The expression distribution of 269 down-regulation genes

表2 差异极显著基因Tab.2 The significant difference genes

表3 差异表达的基因KEGG通路分析Tab.3 The KEGG pathway analysis of different expression genes
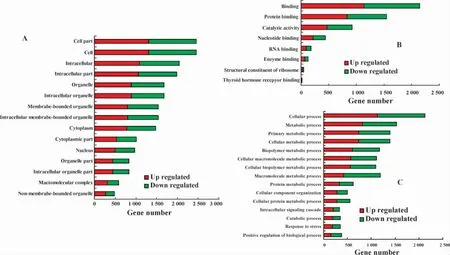
图3 差异基因GO功能富集分析结果Fig.3 The results of differences genes GO function enrichment analysis
2.3 差异基因的GO功能富集分析 本研究将筛选出的差异基因进行了一些列的GO功能富集性分析,包括细胞组成分析(Cellular component)、分子功能分析(Molecular function)、生物过程分析(Biological process)。在进行P值矫正后,取P小于0.05的GO分类条目(term),如表2、图3所示。
2.4 差异基因KEGG代谢通路分析 筛选出差异基因后,为进一步分析这些差异基因在哪些代谢通路中发挥作用,因此我们又做了KEGG代谢通路分析,3097个差异基因能注释到通路中的基因为1508个(人类所有的基因能注释到KEGG通路中的基因数为9424个)取 P值小于0.01,Q值小于0.05的代谢通路作为显著富集的通路,注释到通路中的差异基因数量较多的通路如表3所示。
3 讨论
高通量测序技术的迅猛发展,以及与疾病诊断的加速结合,使得疾病的研究模式发生了重大的转变。以数据化为导向,大规模,工业化的研究模式,极大的提高了疾病的研究效率,革新了人们对于疾病的认识,为疾病的研究,诊断,预防及治疗提供了更为有效的手段。正如科学家预言:使用DNA测序,可以再现致命疾病(肺结核),从人到人的传播过程,快速的识别病原体的来源和活动。这个方法直接向公众宣告了控制感染性疾病暴发流行的新的健康策略。同样本研究也寄希望通过新一代的测序技术,能够找到与肺结核疾病发生相关的基因,为后续的临床研究奠定基础。
本研究中肺结核和健康对照样本经测序比对到参考基因上后,检测到的基因总数分别为12 270,12 244,即存在一部分基因仅在肺结核或者正常样本中表达,如仅在肺结核表达的基因包括(表达量):DEFA4(29.57)、AKR7A2(18.62)、SLC16A6(10.68)、ADRA2B(10.68)、DNAH12(3.56)等,仅在正常样本表达的基因包括(表达量):LOC100286895(103.82)、RASAL2(42)、PMFBP1(31.79)、CYTH3(31.21)、RNF148(29.46)、THAP8(3.5)、CLIC5(3.5)等。如果这些基因被鉴定证实只在肺结核或正常对照中表达,那么这些基因就可被用作肺结核的临床诊断标识。同时造成这种结果的还有一种可能性是某些基因的表达量很低,加上检测灵敏度或样品浓度不够,可能检测不出来。
以 FDR≤0.001,|log2 Ratio|≥1为标准筛选出了3097个差异基因,当将筛选的标准进一步缩小,设定|log2 Ratio|≥4时,发现有16个基因明显明显上调(表2)。其中OSM基因与已经报道的结果相反[13]。IL-15具有与 IL-2相似的作用,可促进T淋巴细胞的增殖分化,诱导多种细胞因子的分泌,对结核患者的保护性免疫机制[14]。其他上调的基因目前没有与肺结核相关的报道,因此关于上调的原因有待于进一步研究。下调的基因269个,远远多于上调的基因,并且其表达量的分布范围也很广泛(图1、2),即可能机体在感染了肺结核以后,大部分基因的表达被抑制,以至于表达量大幅度下降。在肺结核患者中本应正常起作用的基因被抑制,那么很大程度上会对机体的正常代谢造成紊乱,甚至损伤。如果通过一定的手段提高这些基因的表达量,是否会提高机体的免疫力,有利于治疗肺结核,还有待于更深入的研究。在正常对照中表达量最高的三个基因分别为 CXCR4、TWIST2、TYROBP,除了 CXCR4在结核领域研究较多外[15],其他两个基因目前并没有相关的报道,鉴于这三个基因差异极大,很有可能作为候选的诊断标识。
在对差异基因进行GO功能富集性分析时,发现差异基因主要位于细胞内,细胞内相关的细胞器,细胞质等,主要起着链接功能,如蛋白质连接、RNA连接、酶连接、核苷酸连接等,也具有催化活性,少数基因是核糖体的结构组成部分,与甲状腺受体结合,在代谢过程中起作用,包括细胞内代谢、初级代谢、高分子代谢、生物高聚物代谢,还在胞内信号级联中起作用等。每一个GO功能相关的上调基因数基本上多于下调的基因数,有可能在肺结核患者中上调的基因起主要的影响作用。同时这些差异基因也主要在MAPK信号通路、趋化因子信号通路、神经营养蛋白信号通路、T细胞受体信号通路、细胞凋亡等通路中起作用,也与亨廷顿氏舞蹈病、老年痴呆症、帕金森氏病等疾病发生相关。值得一提的是有72个差异基因与癌症的通路(Qvalue=0.0725)有关,当今肺结核合并肺癌的患者越来越多[16],对癌症通路的分析有助于了解肺结核导致肺癌的发生机制。
因为高通量深度测序的结果需要进一步的验证,如qPCR,但是因为样本和经费的原因,未能验证,并且差异基因的相关调控机制尚不清楚,国内也没有相关的报道,有待于更深入的研究,但是,我相信本研究所筛选出来的差异基因能为肺结核领域筛选诊断标识基因,药物作用的靶点等提供参考。
1 Yew W W ,Leung C C.Update in tuberculosis 2007[J].Am J Respir Crit Care Med,2008;177(5):479-485.
2 Ottenhoff T H,Kaufmann S H.Vaccines against tuberculosis:where are we and where do we need to go?[J]PLoS Pathog,2012;8(5):e1002607.
3 Ottenhoff T H,Ellner J J,Kaufmann S H.Ten challenges for TB biomarkers[J].Tuberculosis(Edinb),2012;92(1):17-20.
4 杨 旭,焦 睿,杨 琳et al.基于新一代高通量技术的人类疾病组学研究策略[J].遗传杂志,2011;33(8):829-846.
5 Rosa Rosa J M,Gracia Aznárez F J,Hodges E et al.Deep sequencing of target linkage assay-identified regions in familial breast cancer:methods,analysis pipeline and troubleshooting[J].PLoS One,2010;5(4):e9976.
6 Chou L S,Liu C S J,Boese B et al.DNA sequence capture and enrichment by microarray followed by next-generation sequencing for targeted resequencing:neurofibromatosis type 1 gene as a model[J].Clin Chem,2010;56(1):62-72.
7 Yin L,Liu L,Sun Y et al.High-resolution deep sequencing reveals biodiversity,population structure,and persistence of HIV-1 quasispecies within host ecosystems[J].Retrovirology,2012;9(1):108.
8 Dimitrova Z,Campo D S,Ramachandran S.Evaluation of viral heterogeneity using next-generation sequencing,end-point limiting-dilution and mass spectrometry[J].In Silico Biol,2012;11(5):183-192.
9 Chen X,Yang Q,Zhang M et al.Diagnosis of active tuberculosis in China using an in-house gamma interferon enzyme-linked immunospot assay[J].Clin Vaccine Immunol,2009;16(6):879-884.
10 Hoen P A,Ariyurek Y,Thyqesen H H et al.Deep sequencing-basd expression analysis shows major advances in robustness,resolution and inter-lab portability over five microarray platforms[J].Nucleic Acids Res,2008;36(21):e141.
11 Audic S,Clayerie J M.The significance of digital gene expression profiles[J].Genome Res,1997;7(10):986-995.
12 Benjamini Y,Yekutieli D.The control of the false discovery rate in multiple testing under dependeny[J].The Annals of Statistics,2011;29(4):1165-1188.
13 翟景南,张明霞,张洁云.肺结核患者血浆和胸水中抑瘤素-M检测及临床意义[J].临床肺科,2012;17(4):675-676.
14 杨晓敏,董德琼,李 昶 et al.白细胞介素7和15对肺结核患者Th1/Th2平衡的调节作用[J].中华结核和呼吸杂志,2006;29(6):403-406.
15 Feng L,Li L,Liu Y et al.B lymphocytes that migrate to tuberculous pleural fluid via the SDF-1/CXCR4 axis actively respond to antigens specific for Mycobacterium tuberculosis[J].Eur J Immunol,2011;41(11):3261-3269.
16 明 静,蒋新建.肺结核合并肺癌的临床研究进展[J].现代肿瘤医学,2009;17(12):2426-2428.
